 夜雨聆风
夜雨聆风





文献 | 空间聚集的腺嘌呤碱基编辑器高效纠正少突胶质细胞中的PLP1突变
点击蓝字
关注我们
标题

摘要
少突胶质细胞是中枢神经系统的髓鞘形成细胞,特别容易发生致病性的G-to-A突变,例如导致佩梅病的PLP1A243V突变。佩梅病是一种致命的髓鞘形成不良疾病,目前缺乏有效疗法。虽然腺嘌呤碱基编辑器原则上可以纠正此类突变,但其在少突胶质细胞中的应用受到靶向编辑效率低下的限制。在此,我们开发了一种空间聚集的ABE策略,通过促进tRNA腺苷脱氨酶从细胞质向细胞核的转运来增强编辑。利用基于SunTag的多价招募系统,TadA在基因组靶点处局部富集,实现了体外稳健的编辑。为了在保持高效率和保真度的同时实现体内递送,我们将SpCas9替换为紧凑的eNme2-C Cas9,生成了AAV兼容的变体。值得注意的是,cABE-2.0形成具有液-液相分离特性的动态核斑点,增强了靶向编辑,同时显著降低了全转录组范围的RNA脱靶效应。在功能上,cABE-2.0高效纠正了少突胶质细胞中的PLP1A243V突变,恢复了Plp的亚细胞定位,并挽救了髓鞘形成相关的表型。这些发现表明,空间重组而非提高TadA的内在催化活性,为改善在难以编辑的细胞类型(如少突胶质细胞)中的碱基编辑提供了一条独特的原理,为佩梅病及相关髓鞘疾病的基因治疗提供了机制和技术框架。
关键词: PLP1,佩梅病,少突胶质细胞,COP II,内质网应激,内质网钙衰竭,囊泡运输
引言
少突胶质细胞是中枢神经系统的主要髓鞘形成细胞,特别容易积累G·C到A·T的点突变。当这些突变发生在蛋白质编码区时,它们是少突胶质细胞特异性疾病的主要病因学因素。一个突出的例子是佩梅病,这是一种致命的X连锁脑白质营养不良症,其特征是产生髓鞘的少突胶质细胞严重丧失。该病由蛋白脂蛋白1基因突变引起,导致PLP蛋白错误折叠、异常积累或缺乏。在PLP1突变中,过度表达正常PLP的基因重复产生中度表型,而点突变(如PLP1A243V)约占病例的25%,并导致严重的髓鞘形成不良和早期死亡。目前的疗法是支持性的,缓解症状但不纠正潜在的PLP1突变。因此,在少突胶质细胞中进行直接的基因纠正仍未被探索,使得佩-梅病缺乏治愈性疗法。
腺嘌呤碱基编辑器能以高特异性催化A·T到G·C的替换。然而,它们在蛋白质编码区的效率通常有限且高度可变。这种可变性部分源于编码区复杂的表观遗传景观,包括DNA甲基化和抑制性组蛋白修饰,这些可能使靶点位点对脱氨酶活性产生抵抗。目前增强ABE活性的策略主要依赖于蛋白质工程或编辑器的过表达,这可能会增加转录组范围的脱靶效应,从而限制治疗应用。分裂编辑器架构(如变压器碱基编辑器)允许控制编辑活性的重组,但不改变脱氨酶在基因组靶点的局部浓度。因此,开发通过空间或机制调节同时实现增强的靶向活性和降低脱靶效应的ABE,是精准基因组编辑领域一个关键的未满足需求。
脱氨酶的局部化学计量是ABE活性的关键决定因素。我们假设通过SunTag系统招募TadA可以将编辑活性集中在基因组靶点,提供增强的效率和降低的脱靶效应。首先,TadA招募由抗体-抗原相互作用介导,确保精确定位到靶基因座,并限制转录组中的随机脱氨。其次,该系统的模块化设计允许各个组件保持足够紧凑以便AAV包装,从而实现高效的体内递送。
在本研究中,我们开发了空间聚集的ABE,以在抵抗常规编辑器的少突胶质细胞中实现高效的基因纠正。利用SunTag支架,TadA通过scFv–GCN4相互作用被招募到nSpCas9模块。通过优化TadA化学计量,我们实现了编辑活性的局部富集。为了支持体内应用,我们将SpCas9替换为紧凑的eNme2-C Cas9,产生了cABE-2.0,它在保持高靶向效率和保真度的同时减小了整体编辑器大小。值得注意的是,cABE-2.0形成具有液-液相分离特性的动态核斑点,这些斑点聚集脱氨酶活性,增强靶向DNA编辑,同时显著减少RNA脱靶效应。在功能上,cABE-2.0高效纠正了少突胶质细胞中的Plp1A243V突变,恢复了正确的PLP亚细胞定位,并挽救了髓鞘形成表型。我们的研究结果确立了脱氨酶的空间聚集作为一种独立于蛋白质工程的独特策略,用于在难以编辑的细胞类型中进行精确有效的碱基编辑。
材料与方法
动物
所有操作均按照上海交通大学医学院实验动物科学部批准的方案进行。小鼠饲养于无特定病原体设施中,自由进食。研究中使用的Plp1-CreERT2小鼠(杰克逊实验室,#005975)维持在C57BL/6J背景上,饲养于温度可控的环境中,光照周期为12小时光照:12小时黑暗,自由获取水和啮齿动物饲料。所有小鼠在出生后14天进行基因分型,以确定源自Plp1启动子的Cre序列,使用从消化的尾尖或脚趾中分离的基因组DNA。基因分型通过标准或定制的聚合酶链式反应(PCR)检测进行,基因分型引物见补充表S1。
细胞培养
HEK293T细胞、Neuro-2A细胞和MO3.13细胞
人胚肾(HEK)293T细胞(ATCC CRL-3216)、Neuro-2A细胞(ATCC CCL-131)和MO3.13细胞在含有10%胎牛血清(FBS;Gibco,A5256701)的杜尔贝科改良伊格尔培养基(DMEM)中,于37°C、5% CO2条件下维持培养。
小鼠胚胎干细胞
小鼠胚胎干细胞系(mESCs,E14-Tg2A)如前所述维持。简言之,E14-Tg2A细胞在涂有0.2%明胶(Sigma,9000-70-8)的培养板上,于含有10% FBS(Gibco,10099141)、10% KnockOut血清替代物(KSR;Gibco,A31815-02)、0.1 mM非必需氨基酸(NEAA;Gibco,11140-050)、2 mM GlutaMAX(Gibco,35050-061)、0.1 mM β-巯基乙醇、103单位/ml小鼠白血病抑制因子(mLIF,R&D,8878-LF-100/CF)、1 μM PD0325901(TargetMol,T6189)和3 μM CHIR99021(Tocris BioScience,4423)的KnockOut DMEM(Gibco,10829-018)中,于37°C、5% CO2条件下培养。
CG-4细胞
大鼠少突胶质前体细胞(OPC)系(CG-4)如前所述培养。简言之,CG-4细胞在涂有基质胶(Corning,#354230)的板上,于含有1×N2(BasalMedia,S430J4)、1×B27(BasalMedia,S440J7)、2 mM GlutaMAX、0.1 mM NEAA、20 ng/ml小鼠表皮生长因子(mEGF,Peprotech,315-09)和20 ng/ml血小板衍生生长因子AA(PDGF-AA,Peprotech,315-17)的50% DMEM-F12(Gibco,#C11330500BT)和50% Neurobasal培养基(Gibco,21103-049)中,于37°C、5% CO2条件下维持培养。
原代神经祖细胞
原代神经祖细胞(NPCs)如前所述获取和维持。简言之,原代NPCs从E12.5小鼠皮层中分离,并在涂有基质胶的板上,于含有1×N2、1×B27、2 mM GlutaMAX、0.1 mM NEAA、20 ng/ml mEGF和20 ng/ml碱性成纤维细胞生长因子(bFGF,Peprotech,100-18C)的50% DMEM-F12和50% Neurobasal培养基中,于37°C、5% CO2条件下维持培养。
质粒构建
cABEs的构建中,ABE 8e通过PCR扩增ABE 7.10并引入相关突变获得。质粒CMV-nSpCas9-GCN4-NLS-bGH通过将一个或十个拷贝的GCN4融合到nSpCas9片段的N端或C端生成。然后,将nSpCas9-GCN4克隆到CMV-ABE-NLS-bGH载体(Addgene,#132946)中。质粒U6-sgRNA-CMV-scFV-TadA通过将scFV融合到TadA片段的N端生成,然后将scFV-TadA片段插入U6-sgRNA-CMV-NLS-bGH载体中。用于生成单向导RNA(sgRNA)表达质粒的寡核苷酸(补充表S2)通过退火并通过Golden Gate方法克隆到U6-sgRNA-CMV-NLS-bGH-eGFP载体的BbsI位点中。eNme2-C Cas9序列从CMV-Nme2Cas9-ABE8e(Addgene,#189928)扩增,并亚克隆到CMV-nSpCas9-GCN4-NLS-bGH中替换nSpCas9元件,生成CMV-10×GCN4-linker-eNme2-C-Cas9-NLS-PolyA(5037碱基对,bp)。eNme2-C Cas9的向导RNA支架通过寡核苷酸生成,并生成U6-sgRNA-CMV-scFV-NLS-TadA-PolyA载体(3071 bp),向导RNA通过Golden Gate方法安装到BspQI位点中。为了生成用于慢病毒包装的ABE变体质粒,分别将CMV-ABE-NLS-bGH或U6-sgRNA-scFV-TadA*插入LTR-Tet-o-WPRE-PGK-BSD载体和LTR-EF1α-WPRE-LTR载体中。所有质粒序列均通过DNA测序验证。
为了生成hCD8报告质粒,从小鼠基因组DNA中扩增Tubb3和Omg上游2500 bp以生成启动子Tubb3和Omg(pTubb3和pOmg)。hCD8序列从人类基因组DNA中扩增,并将pTubb3-hCD8或pOmg-hCD8亚克隆到lenti-guide-puro载体中。所有质粒序列均通过DNA测序验证。
PLP1WT或PLP1A243V编码质粒构建
为了生成编码PLP1WT或PLP1A243V的质粒,从人类互补DNA(cDNA)中扩增人PLP1。通过PCR将突变A243V引入PLP1序列,然后将野生型或突变的PLP1序列插入FUW-Tet-o-NLS-PGK-Puro载体中,构建FUW-Tet-o-Flag-PLP1载体。Flag标签在FUW-Tet-o-PLP1质粒中融合到PLP1的N端。所有质粒序列均通过DNA测序验证。
编码cABE变体的AAV构建
为了生成编码cABE变体的AAV,使用质粒AAV-aCamkII-mCherry-P2A-Cre-WPRE-BGH-polyA(Addgene,107312,由Hui Yang惠赠)构建AAV质粒。10×GCN4-eNme2-C Cas9、1×GCN4-eNme2-C Cas9和scFV-TadA的骨架分别从质粒CMV-1×GCN4-eNme2-C-Cas9、CMV-10×GCN4-eNme2-C-Cas9和U6-sgRNA-CMV-scFV-TadA亚克隆。通过分别替换质粒AAV-aCamkII-mCherry-P2A-Cre-WPRE-BGH-polyA中的aCamkII-mCherry-P2A-Cre序列,并连同loxP和loxP 2272元件,构建了质粒AAV-PHP.eB-DIO-1×GCN4-eNme2-C-Cas9(AAV-N1-Cas9)、AAV-PHP.eB-DIO-10×GCN4-eNme2-C-Cas9(AAV-N10-Cas9)和AAV-PHP.eB-scFV-TadA*-DIO-mCherry-U6-sgRNA(AAV-TadA)。U6-sgRNA设计为反向启动以获得更高的转录效率。所有质粒序列均通过DNA测序验证。AAV载体由Brain VTA Biotech包装和纯化。
Sanger测序
将细胞接种在48孔板上,并用表达ABE系统的质粒转染或用携带ABE系统的慢病毒感染。转染或感染12小时后,将细胞更换为250 μL新鲜培养基。转染72小时后,从每孔细胞中分离基因组DNA,并扩增靶序列进行测序分析。用于测序的引物列于补充表S3。
下一代DNA测序和数据分析
下一代DNA测序(NGS)如前所述进行。简言之,使用Phusion Plus DNA聚合酶(Thermo Fisher,F630XL)和含有接头序列的靶位点引物(正向:5′-TTCCCTACACGACGCTCTTCCGATCT-3’,反向:5′-AGTTCAGACGTGTGCTCTTCCGATCT-3’)在5’端(补充表S4)建立NGS DNA扩增子文库。然后,使用含有不同索引序列的引物对上述产物进行第二轮PCR。将得到的扩增子文库等量混合,在Illumina HiSeq平台上进行NGS测序。对于测序数据分析,参考序列设置为从原间隔序列上游10 bp到原间隔序列相邻基序(PAM)序列下游10 bp。使用SHM流程分析原始数据。简言之,使用ea-utils(https://github.com/ExpressionAnalysis/ea-utils)中的fastq-multx工具对双端读段进行解复用。通过BE-Analyzer(http://www.rgenome.net/be-analyzer/#!)分析靶向编辑效率、非预期碱基编辑效率、插入缺失频率和读段计数。碱基编辑值和靶向编辑报告为具有胞嘧啶或腺嘌呤突变的读段数占总比对读段的百分比。
RNA脱靶分析
对于RNA脱靶分析,将细胞接种在48孔板上,并用靶向Site18的表达ABE系统的质粒转染。转染12小时后,将细胞更换为新鲜培养基。处理72小时后,按照标准方案使用TRNzol Universal试剂(TIANGEN,DP424)分离总RNA,然后通过逆转录获得cDNA。使用Phusion Plus DNA聚合酶扩增下一代测序文库,并使用上述方法进行测序。使用SHM流程分析原始数据。通过BE-Analyzer分析RNA脱靶效率。RNA脱靶编辑报告为具有腺嘌呤突变的读段数占总比对读段的百分比。Sanger测序数据通过EditR(1.0.10)(https://moriaritylab.shinyapps.io/editr_v10/)分析。
定量实时PCR
使用TRNzol Universal试剂(TIANGEN,DP424)按照制造商方案从人HEK293T细胞系或小鼠NPC衍生的OPCs中分离总RNA。通过测量A260/A280和A260/A230比值,使用分光光度计(NanoDrop2000,Thermo Fisher Scientific)测定RNA浓度和纯度。使用琼脂糖凝胶电泳评估RNA完整性。对于cDNA合成,根据制造商的说明,使用逆转录试剂盒(Vazyme,R211)将1 μg总RNA在总体积20 μl中进行逆转录。使用SYBR green试剂(Vazyme,Q411)在BIO-RAD CFX Opus 384实时PCR系统上进行定量实时PCR(qRT-PCR),最终反应体积为10 μl,包含各0.2 μM的正向和反向引物。循环参数如下:初始变性95°C 10分钟,随后进行40个循环的变性(95°C,15秒)、退火/延伸(60°C,60秒)。进行65–95°C的熔解曲线分析以确认产物特异性。使用Primer-BLAST设计引物,尽可能跨越外显子-外显子连接处,并由Azenta Life Sciences合成。通过标准曲线(系列10倍稀释)确定扩增效率,在90%–110%之间。引物序列列于补充表S5。使用ΔΔCt方法以GAPDH或Actb作为内参计算相对表达水平。所有反应均进行技术性三重重复,每个实验至少包括三个独立的生物学重复。每次运行均包含无模板和无逆转录对照。数据以平均值±标准差(SD)表示,并使用GraphPad Prism8进行统计分析。此qRT-PCR程序遵循MIQE指南。
细胞转染
对于细胞转染,将HEK293T和Neuro-2A细胞以每孔1×10^5个细胞的密度接种到48孔板中进行转染。使用Lipofectamine LTX(Thermo Fisher,15338100)转染细胞,质粒总量为0.5 μg。对于CG-4或NPC细胞的转染,为1×10^6个细胞制备10 μg质粒混合物,使用Neon电穿孔系统(Invitrogen)在脉冲条件(1350 V,30 ms,单次脉冲)下进行。
携带突变细胞系的建立
通过将ABE系统载体与转移载体psPAX2和编码VSV-G包膜蛋白的pMD2.G载体共转染至汇合度为60%∼70%的HEK293T细胞中来生产慢病毒颗粒。转染后48小时和72小时,从培养基中收集慢病毒颗粒,然后在37°C、5% CO2条件下用于感染HEK293T、CG-4细胞或原代NPCs。细胞在37°C、5% CO2条件下用5 μg/ml嘌呤霉素筛选至少7天。通过测序验证报告基因表达的阳性细胞群,并用于后续实验。
小鼠胚胎干细胞的体外分化
神经元分化
神经元分化按文献所述进行。简言之,为诱导神经元,首先将mESCs分化为NPCs。将1×10^5个mESCs接种到非粘附性培养板中,使用含有10% KSR血清、0.1 mM NEAA、0.1 mM β-巯基乙醇和2 mM GlutaMAX的Knockout DMEM培养基。每天更换培养基,5天后添加5 μM视黄酸。然后将神经球解离并接种到基质胶包被的培养板上,使用含有1×N2、不含维生素A的1×B27(Gibco,12587010)、0.075%牛血清白蛋白(BSA)、0.1 mM NEAA、2 mM GlutaMAX、20 ng/ml bFGF、20 ng/ml mEGF的50% Neural basal培养基和50% DMEM/F12培养基,如前所述。培养和迁移3∼4天后,将NPCs接种到基质胶包被的培养板上,使用含有1×N2、不含维生素A的1×B27、0.1 mM NEAA、2 mM GlutaMAX、0.075% BSA、200 μM抗坏血酸、2 μM db-cAMP(TOCRIS,1141)、25 ng/ml脑源性神经营养因子(BDNF,Peprotech,450-02)、25 ng/ml神经营养因子-3(NT3,Peprotech,450-03)、50 ng/ml胶质细胞源性神经营养因子(GDNF,Peprotech,450–10)的50% Neural basal培养基和50% DMEM/F12培养基,在37°C、5% CO2条件下诱导神经元。
少突胶质细胞分化
OPC分化如前所述进行。将NPCs接种到基质胶包被的培养板上,使用含有1×N2、不含维生素A的1×B27、0.1 mM NEAA、2 mM GlutaMAX、0.1 mM β-巯基乙醇、200 ng/ml Shh(Primegene,601-19)、20 ng/ml bFGF、20 ng/ml PDGF-AA的50% Neural basal培养基和50% DMEM/F12培养基,在37°C、5% CO2条件下每两天更换一次培养基。OPC诱导5天后,将细胞更换为含有1×N2、不含维生素A的1×B27、0.1 mM NEAA、2 mM GlutaMAX、0.1 mM β-巯基乙醇、200 ng/ml Shh、100 nM LDN193189(Collagen Technology,#C5361-2)、100 ng/ml胰岛素样生长因子1(IGF-1,Dakewe. 6146513)、10 mM环磷酸腺苷(TOCRIS,1141)、10 ng/ml NT3、40 ng/ml三碘甲状腺原氨酸(T3,APExBIO,C6407)的50% Neural basal培养基和50% DMEM/F12培养基,在37°C、5% CO2条件下培养5–6天,以诱导少突胶质细胞分化和成熟。
体外髓鞘形成实验
CG-4来源的OLs与诱导神经元的共培养方法根据已报道的方案进行了修改。简言之,将CG-4细胞加载到诱导神经元上,使用50%神经元培养基和50% CG-4分化培养基,在37°C、5% CO2条件下共培养7天。CG-4分化培养基含有1×N2、1×B27、2 mM GlutaMAX、0.1 mM NEAA、5 μg/ml胰岛素、50 μg/ml转铁蛋白(BasalMedia,S450J7)、50 mg/ml睫状神经营养因子(CNTF,MCE,HY-P7145)、60 mg/ml孕酮、16.1 μg/ml腐胺、5 μg/ml硒化钠和400 ng/ml T3(APExBIO,C6407)。然后固定细胞并通过免疫荧光进行分析。
流式细胞术和细胞分选
对于hCD8+细胞类型特异性分离,收集细胞,洗涤,并在含有1% FBS的冰冷磷酸盐缓冲盐水(PBS)中调整至浓度为1×10^6个细胞/ml。用稀释的一抗APC-抗CD8抗体(Abcam,ab93278)在4°C下染色和孵育30分钟。染色后,通过400 × g离心5分钟洗涤细胞三次,并重悬于500 μl至1 ml冰冷的PBS中。将细胞在冰上避光保存,并使用Aria II细胞分选仪(BD Biosciences)进行分选,数据使用FlowJo 10.8.1进行分析。
为了分离AAV感染的细胞进行测序,取出小鼠大脑并进行解剖:用手术分离大脑半球,用弯曲的刮匙分离新皮质(包括胼胝体)。所有步骤均在冰上进行。将组织置于荧光解剖显微镜下以识别转染区域,并在解剖培养基(含有1% GlutaMAX、1%抗生素、1 mg/ml木瓜蛋白酶、5.5 mM L-半胱氨酸、1.1 mM乙二胺四乙酸(EDTA)、0.067 mM β-巯基乙醇、50 mM Tris–HCl的Neurobasal培养基)中于37°C孵育30分钟以解离成单细胞。收集细胞,洗涤,并在含有1% FBS的冰冷PBS中调整至浓度为1×10^6个细胞/ml。在Aria II细胞分选仪(BD Biosciences)上进行流式细胞术,数据使用FlowJo 10.8.1进行分析。
通过侧向散射面积与正向散射面积(SSC-A与FSC-A)圈定活细胞。通过正向散射高度与正向散射面积(FSC-H与FSC-A)选择单细胞。针对模拟转染对照圈定荧光阳性群体。
1,6-己二醇处理
活细胞用1,6-己二醇(1,6-HD;MACKLIN,C16670511)处理如前所述。简言之,将HEK293T细胞培养在置于24孔板中的玻璃盖玻片上,并用指定的ABE构建体转染。转染48小时后,将盖玻片转移至新鲜培养基中进行活细胞成像。对于1,6-HD处理,在水中制备15%(w/v)的1,6-HD储备液,并在预热的培养基中稀释至终浓度7.5%。将培养基更换为含1,6-HD的培养基,并立即以2秒间隔对细胞进行连续延时荧光成像。在1,6-HD添加、洗脱和恢复过程中,成像持续不间断进行。
免疫细胞化学
免疫细胞化学如前所述进行。用4%多聚甲醛(PFA)的PBS溶液固定细胞。固定后,用含有0.2% Triton X-100(Sigma,T8787)的PBS溶液透化细胞,然后在含有7.5% BSA的PBS溶液中封闭。细胞在4°C下用以下稀释于封闭液中的一抗染色过夜:大鼠抗MBP(Abcam,ab7349,1:500)、小鼠抗Flag(Sigma,F1804,1:500)、兔抗NF(CST,2837S,1:500)。对于二抗免疫染色,使用Alexa Fluor抗体(Thermo Fisher),浓度为1 μg/ml,并使用4′,6-二脒基-2-苯基吲哚(DAPI,Sigma,D8417,100 ng/ml)识别细胞核。使用尼康ECLIPSE Ti2荧光显微镜捕获图像,并使用ImageJ软件进行分析。
免疫组织化学
免疫组织化学按文献所述进行。用异氟烷麻醉小鼠,经颅灌注PBS,随后灌注含4% PFA的PBS溶液进行安乐死。收集组织,置于含4% PFA的PBS溶液中,于4°C过夜。样品切片厚度为20 μm。切片用PBS洗涤,并在含有5%正常驴血清(Jackson Laboratories,017-000-121)和0.3% Triton X-100的抗体溶液中孵育过夜。使用以下抗体以指定浓度或稀释度进行染色:兔抗Plp(CST,28702,1:200)。使用Alexa Fluor抗体(Thermo Fisher)进行二抗免疫染色,浓度为1 μg/ml。使用DAPI(100 ng/ml)识别细胞核。使用尼康ECLIPSE Ti2荧光显微镜捕获染色切片,并使用ImageJ软件进行分析。所有计数和定量均在盲法下进行。
蛋白质印迹
蛋白质印迹按文献所述进行。向每个样品中加入由RIPA缓冲液(Sigma,R0278)和苯甲基磺酰氟(PMSF)组成的蛋白裂解缓冲液。裂解液在4°C下以17,000 × g离心15分钟进行分离。使用Pierce BCA蛋白测定试剂盒(Thermo Scientific,23225)生成二喹啉甲酸(BCA)标准曲线,用于将样品标准化至等效蛋白浓度。使用一步法聚丙烯酰胺凝胶电泳(PAGE)凝胶快速制备试剂盒(Vazyme,E303-01)在10% Bis–Tris蛋白凝胶上等量上样样品,然后电转移到聚偏二氟乙烯(PVDF)膜(Millipore,IPVH00010)上。膜用含5%脱脂牛奶的TBS-T封闭1小时,然后在4°C下与兔抗Plp抗体(CST,28702,1:1000)或小鼠抗β-肌动蛋白抗体(ABclonal,AC026,1:5000)杂交过夜。然后用TBS-T洗涤印迹,并在山羊抗小鼠辣根过氧化物酶(HRP;CST,7076,1:10,000)、山羊抗兔HRP(CST,7077,1:10,000)中孵育。所有二抗在室温下孵育2小时。使用ImageQuant LAS 4000 mini(GE Healthcare Bio-Sciences AB)分析印迹。
他莫昔芬给药
将他莫昔芬(TargetMol,T6906)溶解于玉米油中,浓度为30 mg/ml。所有小鼠通过口服灌胃给予他莫昔芬溶液,剂量为100 mg/kg/天,连续7天以诱导重组。
立体定位注射
注射前,将AAV用0.9% NaCl稀释至5 μL。用异氟烷(4%诱导,100% O2中1.5%–2%维持)麻醉8周龄C57BL/6小鼠,并将其置于带有加热垫的立体定位框架(Reward)中,以维持体温在37°C。用碘酒和乙醇消毒头皮,并沿中线切开以暴露颅骨。在目标坐标(前-后,-1.00 mm;内-外,±1.04 mm;背-腹,距前囟-1.38 mm)处钻一个小颅骨开窗(直径约0.5 mm)。将AAV(3×10^12 vg/ml)装入汉密尔顿注射器(33号针头),并使用微量注射泵(Reward)以250 nl/min的速度输注(每个位点总计5 μL)。注射后针头在原位停留10分钟以防止回流。用5-0可吸收缝线缝合头皮,并局部涂抹抗生素软膏。监测小鼠直至完全清醒,并在术后48小时内给予美洛昔康(5 mg/kg)镇痛。4周后,对大脑进行切片并染色以检测靶蛋白表达。通过转棒实验评估运动功能以验证生理影响。所用AAV的具体载体滴度如下:AAV-PHP.eB-DIO-10×GCN4-eNme2-C-Cas9:6.45×10^12 vg/mL;AAV-PHP.eB-scFV-TadA*-DIO-mCherry-U6-sgRNA:5.83×10^12 vg/mL;AAV-PHP.eB-DIO-1×GCN4-eNme2-C-Cas9:5.18×10^12 vg/mL;AAV-PHP.eB-DIO-mCherry:5.08×10^12 vg/mL。
转棒实验
实验前,如文献所述,需要对小鼠进行几次转棒训练,使其适应设备并确保测试期间的舒适度。使用加速转棒系统,旋转棒直径为3厘米,分为多个通道以同时测试多只小鼠。测试前,让小鼠在测试室适应≥30分钟。按照为期2天的预训练方案训练小鼠:第1天,将小鼠置于恒定速度(4 rpm)的转棒上60秒,重复3次,每次间隔10分钟。第2天,以8 rpm的速度测试小鼠120秒(3次试验,间隔10分钟)。对于加速转棒测试,速度在5分钟内从4 rpm线性增加到40 rpm(最长持续300秒)。连续3天每天进行3次试验,间隔30分钟。通过红外传感器自动记录跌落潜伏期(秒)。在300秒时仍停留在转棒上的小鼠被移除并记为300秒。对于恒定速度转棒测试,速度固定为20 rpm以评估运动耐力。小鼠进行2次试验,最长180秒,间隔1小时。排除紧抓转棒但不行走(>2圈)的小鼠。
统计学分析
使用GraphPad Prism 8(GraphPad Software)进行统计分析。除非另有说明,数值以平均值±标准差表示,并基于至少三个独立的生物学重复进行量化。未使用统计方法预先确定样本量。对于两组比较,使用双尾非配对Student’s t检验确定统计学显著性。对于多组比较,进行单因素方差分析(ANOVA)。具体的统计方法在每个图例中描述。
结果
cABEs的建立
为了建立cABEs,我们比较了基于nSpCas9的ABE8e及其整合到SunTag系统中的分裂衍生物的编辑效率(图1A)。模块化的nSp-ABE8e在SunTag支架上组装,其中nSpCas9与GCN4融合(nSpCas9模块),TadA与scFv融合(TadA模块)。考虑到TadA相对于nSpCas9的空间位置可能影响编辑活性,我们对GCN4进行工程改造,使其连接到nSpCas9的C端(C1-nSp-ABE8e)或N端(N1-nSp-ABE8e)。通过多西环素(Dox)诱导模块的表达,从而通过GCN4–scFv相互作用实现可控的编辑活性重组。作为比较,单质粒(ABE8e)和双质粒(DPS-nSp-ABE8e,DPS)形式的ABE8e作为对照。将这些变体的编码质粒转导到HEK293T细胞中,并评估编辑效率(补充图S1A和B)。虽然DPS表现出与ABE8e相当的活性,但初始分裂变体(C1-和N1-nSp-ABE8e)显示出略微降低的效率,可能是由于重组不理想。
然后,我们通过改变GCN4拷贝数来调节TadA化学计量,发现在nSpCas9:TadA比例为1:10时达到最佳活性。与ABE8e相比,C10-nSp-ABE8e的编辑活性增加了1.96倍,N10-nSp-ABE8e增加了2.12倍(补充图S1C和D),而nSpCas9、TadA或向导RNA(gRNA)的表达没有变化(补充图S1E)。在这些变体中,N10-nSp-ABE8e(cABE-1.0)表现出最高的编辑效率,这在包含55个位点的10个基因组位点上得到了验证(补充图S2A)。平均而言,cABE-1.0相对于ABE8e将编辑效率提高了3.46倍,突显了空间集中招募TadA的优势(图1B和C,以及补充图S2B)。
AAV兼容的cABE-2.0的开发
为了优化cABE-1.0以用于AAV包装,我们用紧凑的eNme2-C Cas9 替换了nSpCas9,并通过在eNme2-C Cas9的N端整合1个或10个GCN4拷贝生成了N1-eNme2-C-ABE8e和N10-eNme2-C-ABE8e(图1A)。这一修改将编辑器总大小减少了约18%,使其与AAV载体兼容。值得注意的是,N10-eNme2-C-ABE8e(cABE-2.0)在11个基因组位点(35个编辑位点)上的编辑活性比eNme2-C-ABE8e提高了3.09倍,并且在不同的PAM上优于cABE-1.0(图1D–G,以及补充图S2B和C)。
为了评估疾病靶向潜力,我们查询了ClinVar以评估对人类基因组中致病性腺嘌呤位点的覆盖情况。cABE-1.0(基于nSpCas9)可以靶向这些位点的52.3%,而cABE-2.0(基于eNme2-C Cas9)将覆盖率扩展到73.6%(补充图S2D)。增强的编辑效率和AAV兼容性的结合,使cABE-2.0成为体内基因纠正和潜在治疗应用的优越平台。
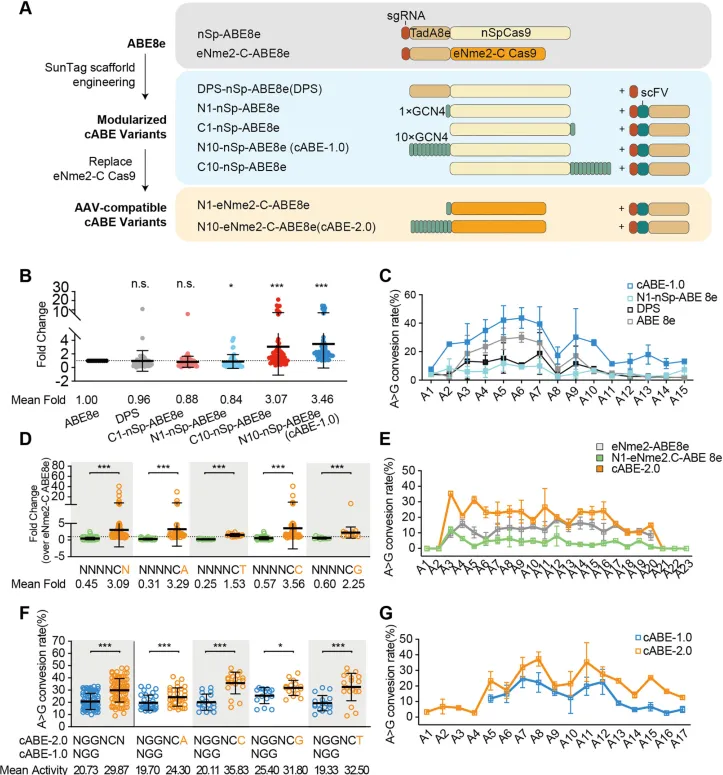
图1 |图1.空间聚集的腺嘌呤碱基编辑器(cABEs)的开发与优化。
(A) nSp-ABE8e、eNme2-CABE8e以及整合了SunTag系统的cABE变体的示意图。nSp-ABE8e被模块化为TadA*-nSpCas9和sgRNA模块,以生成双质粒系统(DPS)nSp-ABE8e。cABE变体通过在nSp-Cas9或eNme2-CCas9的N端或C端融合GCN4肽段,并将scFV融合到TadA*上,从而实现多价的SunTag介导的招募。每种cABE的详细结构如(A)所示。
(B) nSp-cABE变体相对于nSp-ABE8e(ABE8e)的编辑活性总倍数变化(n=3个独立的生物学重复)。
(C) ABE8e、N1-nSp-ABE8e、DPS和cABE-1.0的编辑窗口(n=3个独立的生物学重复)。
(D) 基于eNme2-C的cABE变体相对于eNme2-CABE8e的编辑活性总倍数变化(n=3个独立的生物学重复)。
(E) eNme2-CABE8e、N1-eNme2-C-ABE8e和cABE-2.0的编辑窗口(n=3个独立的生物学重复)。
(F) cABE-1.0和cABE-2.0在HEK293T细胞中10个PAM匹配的N4CN/NGG位点的编辑效率。最左侧一列代表所有10个位点的汇总数据,后续列按特定PAM分组(n=3个独立的生物学重复)。
(G) cABE-1.0和cABE-2.0的编辑窗口(n=3个独立的生物学重复)。
(B-G)图中的数据以平均值±标准差表示。P值使用单因素方差分析(B)或双尾Student’st检验(D,F)计算。*P<.05,**P<.01,***P<.001。
cABE-2.0能够在染色质抵抗位点实现稳健编辑
编码区通常受到复杂表观遗传景观的限制,阻碍碱基编辑器活性。为了评估cABE在此类位点的性能,我们整合了HEK293T ChIP-seq数据和ClinVar G·C到A·T突变条目,并选择了20个不同染色体上的神经相关位点。这些包括10个常染色质位点(47个位点),标记为H3K4me3,以及10个异染色质位点(53个位点),富含H3K9me3和H3K27me3(补充图S3A和B)。Sanger测序显示,cABE-2.0在常染色质位点实现了高达67.0%的转换(平均44.3%),比ABE8e(26.9%)提高了3.27倍。在异染色质位点,cABE-2.0达到高达53.0%的效率(平均37.3%),比ABE8e(24.8%)提高了4.62倍(图2A-C)。这些结果表明,cABE-2.0有效克服了染色质介导的障碍,能够在常染色质和异染色质位点实现稳健编辑。
cABE-2.0克服DNA甲基化障碍
DNA甲基化是限制碱基编辑效率的另一个主要决定因素。我们选择了12个基因组位点,包含19个高甲基化位点(MS ≥ 50%)和11个低甲基化位点(12% ≤ MS ≤ 50%)(补充图S3C–F)。Sanger测序显示,cABE-1.0在高甲基化和低甲基化位点的平均编辑率分别为24.67%和22.14%,而cABE-2.0达到34.05%和27.74%(图2D)。相对于ABE8e,cABE-1.0在高甲基化和低甲基化背景下分别将编辑提高了3.83倍和4.95倍,而cABE-2.0分别实现了5.92倍和7.40倍的增强(图2E和F)。这些结果强调了cABE-2.0克服DNA甲基化介导障碍的能力,突显了其在多样化表观遗传景观中的稳健性。
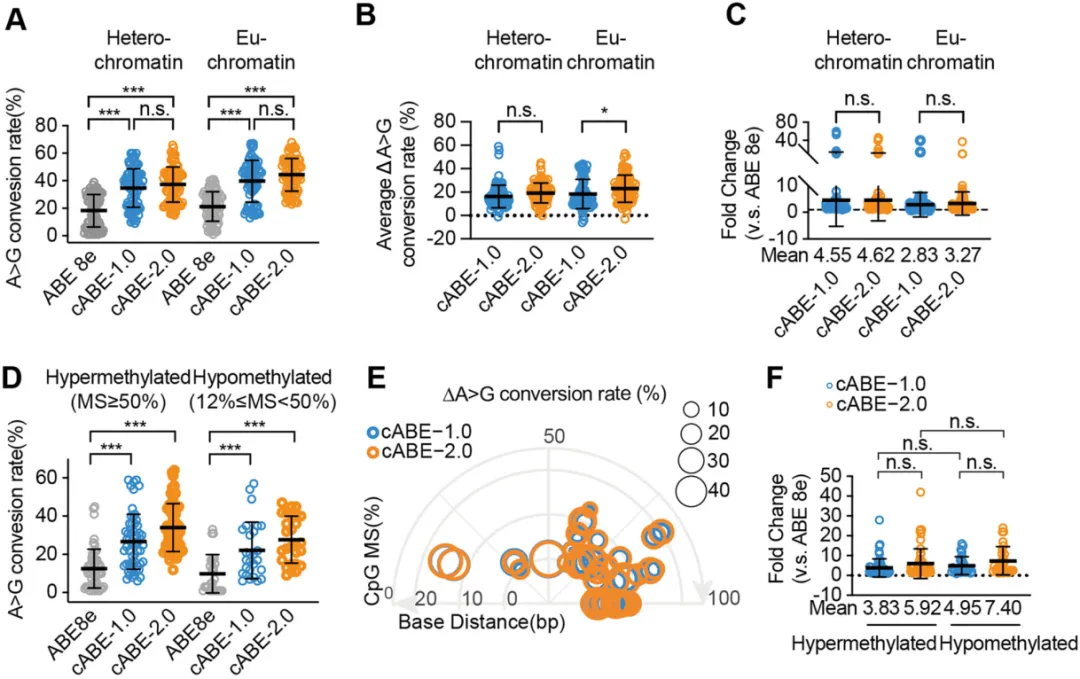
图2 |cABEs在抵抗性染色质环境中的编辑效率。
(A–C)ABE8e、cABE-1.0和cABE-2.0在常染色质位点和异染色质位点的汇总编辑效率(A)、绝对变化(B)和倍数变化(C)。
(D)ABE8e、cABE-1.0和cABE-2.0在高甲基化(MS≥50%)和低甲基化(12%≤MS<50%)位点的汇总编辑效率。
(E)cABEs相对于ABE8e的平均编辑绝对变化的极坐标图,角度代表甲基化状态,直径代表碱基位置。
(F)cABE-1.0和cABE-2.0在高甲基化和低甲基化位点相对于ABE8e的汇总倍数变化。
所有图n=3个独立的生物学重复。所有图的数据以平均值±标准差表示。P值使用单因素方差分析(A,D)或双尾Student’st检验(B,C,F)计算。*P<.05,**P<.01,***P<.001。
cABEs保持低gRNA依赖性DNA脱靶编辑
基因组DNA的脱靶编辑是治疗性ABE的主要障碍。为了在生理相关背景下评估gRNA依赖性脱靶编辑,我们在人OPC系MO3.13中评估了cABEs。分析了两个已确立的靶向位点(Site-m1和Site-m2)以及16个与SpCas9和eNme2-C Cas9兼容的额外潜在脱靶位点(图3A和B,以及补充表S6)。cABE-2.0在Site-m1和Site-m2分别实现了34.72%和29.29%的靶向编辑,显著高于ABE8e(3.46%和12.52%)和cABE-1.0(26.25%和26.30%)(图3B)。在16个潜在脱靶位点中,所有变体在大多数位点的编辑率保持在0.50%–0.65%以下,只有三个例外显示出适度升高的脱靶活性(图3C和D)。重要的是,平均脱靶编辑效率在ABE8e(1.61%)、cABE-1.0(1.61%)和cABE-2.0(1.56%)之间具有可比性(图3E),表明cABEs保持了低gRNA依赖性基因组脱靶谱。
cABEs保持低gRNA非依赖性DNA脱靶编辑
为了评估gRNA非依赖性脱靶活性,我们使用了由金黄色葡萄球菌Cas9(SaCas9)生成的R环底物,为脱氨酶活性提供单链DNA(图3F)。下一代测序(NGS)分析显示,相对于ABE8e,cABEs的脱靶编辑略有增加。然而,在所有R环位点上,总体编辑率保持在1.5%以下(图3G和H)。这些结果表明,cABEs在实现显著增强的靶向编辑效率的同时,保持了良好的基因组安全性。
cABEs降低RNA脱靶编辑
已知ABEs会诱导RNA脱靶编辑,这是治疗应用中的一个主要安全问题(图3I)。为了评估cABEs是否改变RNA底物的可及性,用靶向Site18的gRNA与ABE8e、cABE-1.0或cABE-2.0共转染HEK293T细胞。在跨越16条染色体的22个转录本上分析了RNA脱靶编辑,涵盖了109个RNA脱靶热点(补充表S7)。与ABE8e相比,两种cABEs均显著且一致地抑制了RNA脱靶事件(图3J)。ABE8e的平均RNA脱靶编辑率为2.52%,而cABE-1.0和cABE-2.0分别将其降低至0.21%和0.13%(图3K),这表明TadA*的空间集中有效地限制了RNA脱靶脱氨。
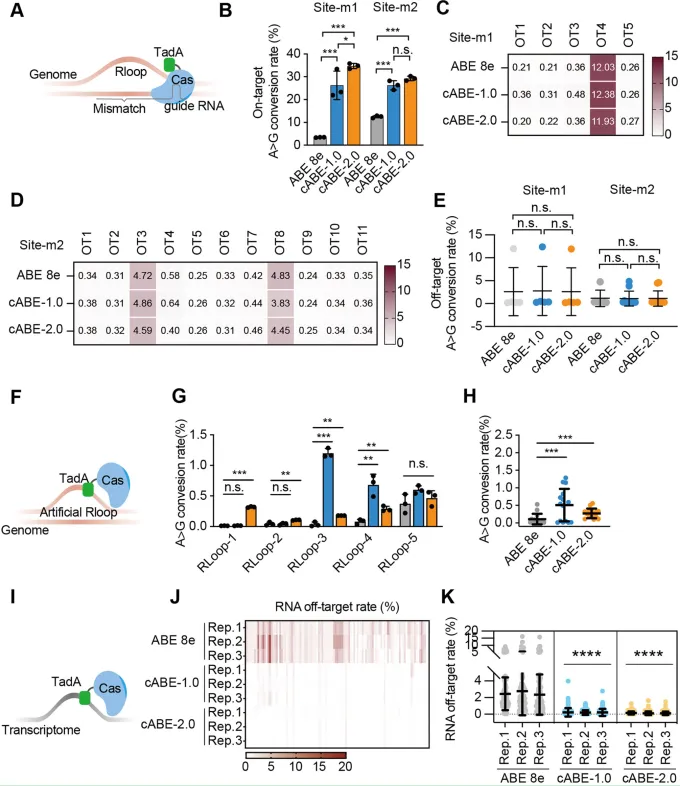
图3 |cABEs在基因组和转录水平降低脱靶效应。
(A) 在Site-m1和Site-m2进行Cas9依赖性DNA脱靶评估的示意图。
(B) ABE8e、cABE-1.0和cABE-2.0在Site-m1和Site-m2的靶向编辑效率(n=3个独立的生物学重复)。
(C,D) ABE8e、cABE-1.0和cABE-2.0在Site-m1(C)和Site-m2(D)的脱靶编辑效率(n=3个独立的生物学重复)。
(E) 在Site-m1的5个预测脱靶位点和Site-m2的11个预测脱靶位点的汇总脱靶编辑效率(n=3个独立的生物学重复)。
(F) 使用失活的SaCas9生成的Rloop-1∼5评估Cas9非依赖性DNA脱靶效应的示意图。
(G) ABE8e、cABE-1.0和cABE-2.0在Rloop-1∼5底物上的脱靶编辑效率(n=3个独立的生物学重复)。
(H) ABE8e、cABE-1.0和cABE-2.0在Rloop-1∼5上的汇总Cas9非依赖性DNA脱靶效率(n=3个独立的生物学重复)。
(I) 游离脱氨酶诱导ABERNA脱靶效应的示意图。
(J) ABE8e、cABE-1.0和cABE-2.0在22个转录本上的脱靶编辑效率(n=3个独立的生物学重复)。22个转录本的109个腺嘌呤的详细信息列于补充表S7。
(K) ABE8e、cABE-1.0和cABE-2.0的汇总RNA脱靶效率(n=3个独立的生物学重复)。
数据以平均值±标准差表示(B,E,G,H,K)。P值使用单因素方差分析(B,E,G,H,K)计算。*P<.05,**P<.01,***P<.001。
cABEs促进TadA的核转位
为了研究cABEs增强靶向DNA编辑和降低RNA脱靶活性的机制,我们假设SunTag介导的多价招募驱动了TadA*的核转位,从而将脱氨酶集中在靶位点,同时限制其在细胞质中的扩散。用分别带有HA和GFP标签的Cas9和scFv-TadA*转染HEK293T细胞,并通过靶向端粒的gRNA引导。在表达ABE8e的细胞中,两个组分均表现出主要的弥散性核质定位(图4A和B,以及补充图S4A)。相比之下,cABEs主要局限于细胞核并形成离散的点状结构,定量荧光分析证实了Cas9和TadA*在这些结构内的共定位(图4C和D,以及补充图S4B和C)。这些结果表明,cABEs有效地将TadA*隔离到细胞核中,为增强靶向编辑和减少RNA可及性提供了空间基础。
cABEs形成具有LLPS特性的动态核斑点
为了检查核斑点的生物物理性质,用1,6-己二醇处理活细胞,它会破坏LLPS特征的弱疏水相互作用。cABE-1.0和cABE-2.0形成的斑点在1,6-己二醇处理后迅速溶解,并在洗脱后分别在6.23秒和7.17秒内重新形成(图4A和E,以及补充图S5A和B),表明其具有高动态性和类液体的可逆性。相比之下,N端20×和40×GCN4融合体显示出最小的荧光衰减且未能恢复,表明是刚性组装(补充图S5A和B)。定量分析显示,每个细胞核的斑点丰度在10×SunTag构型时达到峰值,在更高拷贝数时下降(图4F和补充图S5C)。这种非单调模式与之前的SunTag研究一致,反映了由于过度多价性导致的分子流动性降低。相应地,斑点丰度的下降与编辑效率的降低相关(图4G),支持了动态的类LLPS组装是cABEs增强活性的基础。
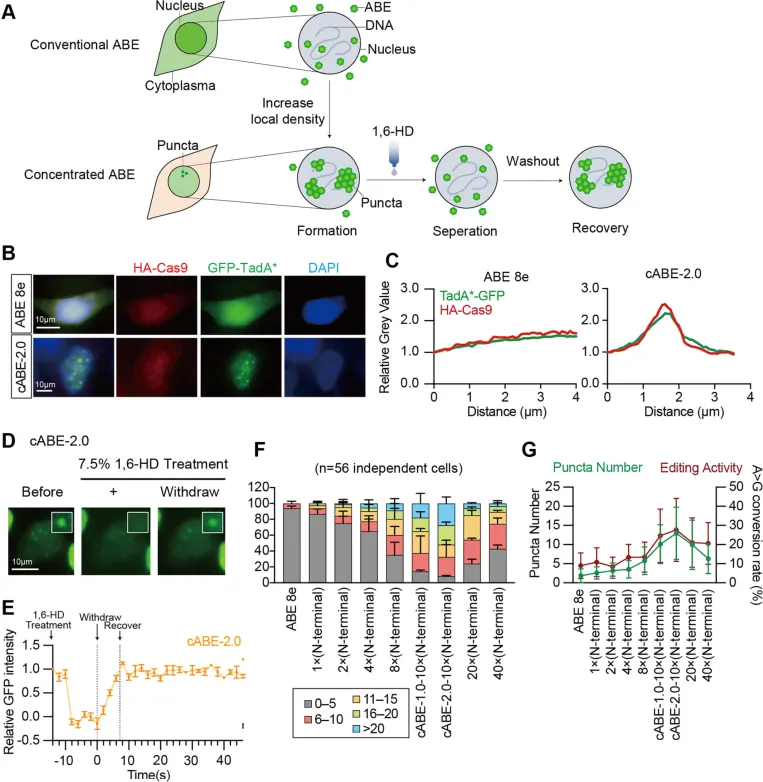
图4 |cABEs的空间聚集通过形成类LLPS核斑点减少胞质脱氨酶分布。
(A) 示意图说明ABE变体的亚细胞定位模式以及斑点在存在或不存在1,6-HD情况下的动态行为。
(B) ABE8e和cABE-2.0在HEK293T细胞中的代表性图像,显示eGFP融合的scFv-TadA*(绿色)、HA标记的Cas9(红色)和DAPI(蓝色)。比例尺,10μm。
(C) 相对荧光强度的线扫描分析,证明HA标记的Cas9和TadA*-GFP在cABE-2.0形成的核斑点内的空间共定位。
(D) 代表性图像显示cABE-2.0在7.5%1,6-HD处理前后形成的TadA*-GFP斑点。一个代表性斑点用白色方框标出。比例尺,10μm。
(E) 1,6-HD处理后荧光恢复的量化,显示(D)中斑点区域内随时间变化的相对GFP强度。在指示的时间点添加或洗脱1,6-HD。平均恢复时间根据荧光曲线计算。数据以平均值±标准误表示。n=每组5个独立细胞中的5个独立斑点。
(F) 每种ABE变体每个细胞的斑点数量分布。根据斑点数量对细胞进行分类,并归一化到分析细胞的总数。n=每组56个独立细胞,来自三个独立的生物学实验。数据以平均值±标准差表示。
(G) 不同ABE变体之间每个细胞的平均斑点数量(左,绿色)与在Site18位点的相应基因组编辑效率(右,红色)的比较。每组数据以平均值±标准差表示。
cABEs保持低gRNA依赖性DNA脱靶编辑
为了评估cABE性能的普适性,我们在多个人类细胞系(HeLa、SW1116和H1299)中评估了在Site19的编辑活性(补充图S6A)。在HeLa细胞中,ABE8e在A6位置达到最高32.17%的编辑效率,但在相邻位点(A8)效率下降,在远端位置(A12,A14)可忽略不计。相比之下,cABE-2.0在HeLa、SW1116和H1299细胞中分别比ABE8e持续提高了4.46倍、6.56倍和2.66倍的编辑效率,而cABE-1.0仅实现了更温和的2.64倍、3.94倍和1.50倍的提升(补充图S6B-E)。这些结果表明,cABE-2.0在不同的人类细胞类型中稳健地增强了靶向编辑。
cABE-2.0改善小鼠神经细胞的编辑
接下来,我们评估了在小鼠Neuro-2A神经母细胞瘤细胞(神经谱系应用的模型)中的编辑。cABE-2.0实现了15.83%的编辑,比ABE8e提高了4.62倍,而cABE-1.0仅提供了1.90倍的提升(补充图S6F-H)。为了将此分析扩展到有丝分裂后的神经细胞,我们靶向了源自小鼠胚胎NPCs的神经元和OLs中的Rosa26位点,使用细胞类型特异性报告基因(用于神经元的pTubb3-humanCD8,用于OLs的pOmg-humanCD8)以实现基于FACS的纯化(补充图S7A–D)。在编辑窗口内,ABE8e在Neuro-2A中达到33.00%,在神经元中达到18.56%,在OLs中仅为5.44%,突显了OLs对基因组编辑的内在抵抗性(补充图S7E–G)。cABE-2.0在所有三种细胞类型中均显著提高了编辑效率,在Neuro-2A中达到53.33%(1.62倍),在神经元中达到38.78%(2.09倍),在OLs中达到15.11%(2.78倍)(补充图S7H)。这种在更难编辑的细胞类型中逐步增强的效果,突显了cABE-2.0在人类和神经谱系中进行高效基因组编辑的广泛适用性。
cABE-2.0能够在OPCs中高效编辑疾病相关位点
OL特异性基因(包括MOG、OLIG1和MOBP)的突变是遗传性脑白质营养不良和脱髓鞘疾病的基础。为了在疾病相关背景下评估cABEs,我们使用三种OPC模型(人MO3.13细胞、小鼠原代NPC来源的OPCs和大鼠CG-4OPCs)靶向这些基因中的致病性或疾病相关位点(图5A)。我们比较了基于SpCas9的传统ABE8e与cABE-1.0,以及基于eNme2-CCas9的eNme2-CABE8e与cABE-2.0。
在所有三个物种和靶基因中,cABE-1.0的表现始终优于ABE8e,平均编辑效率为28.26%,而ABE8e为4.46%,相当于提高了6.34倍(图5A-C和补充图S7I)。cABE-1.0的编辑效率范围从15.33%(人OLIG1)到52.67%(小鼠Mog),在跨物种的MOG/Mog位点表现出特别强的活性(49.33%–52.67%)。cABE-2.0进一步增强了编辑,平均效率为41.59%,而eNme2-CABE8e为16.50%(提高了2.52倍)(图5A-C和补充图S7I)。值得注意的是,cABE-2.0在人和小鼠OPCs的MOG/Mog位点编辑效率超过70%,在人MOBP位点保持>50%的效率,甚至在跨物种更难编辑的OLIG1/Olig1位点也实现了20.33%–29.16%的编辑。
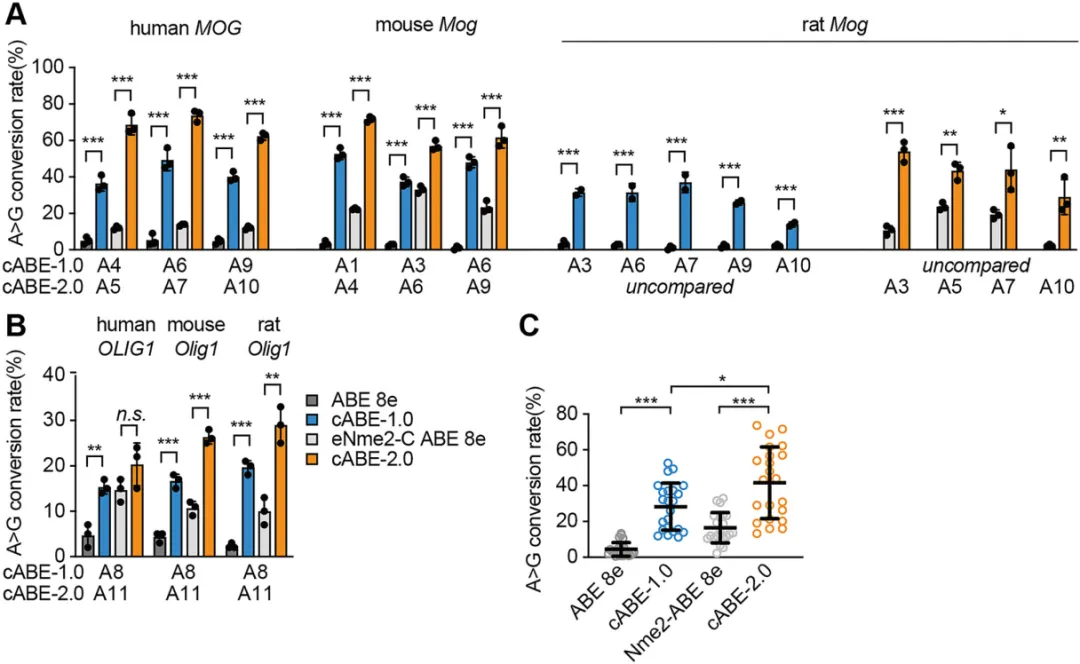
图5 |cABEs在少突胶质细胞谱系细胞中增强的编辑活性。
(A,B) ABE8e、cABE-1.0、eNme2-CABE8e和cABE-2.0在不同物种的少突胶质前体细胞(OPCs)中MOG/Mog(A)和OLIG1/Olig1(B)位点的编辑效率(n=3个独立的生物学重复)。
(C) 跨OPCs的汇总编辑效率。数据以平均值±标准差表示。P值使用单因素方差分析(C)或双尾Student’st检验(A,B)计算。*P<.05,**P<.01,***P<.001。(关于图例中颜色的解释,请读者参考本文的网络版。)
cABE-2.0纠正PMD相关的PLP1突变
PLP1中的c.725C>T点突变(p.Ala243Val;PLP1A243V)导致PLP蛋白从质膜错误定位到核周区室,引发少突胶质细胞凋亡和严重的髓鞘形成不良,是PMD最具侵袭性的形式之一(图6A和补充图S8A和B)。为了评估cABE-2.0是否能纠正这种致病性突变,我们生成了表达Flag标签的人PLPWT或PLPA243V的稳定大鼠CG-4细胞系。用靶向突变位点的ABE变体转导细胞。Sanger测序显示,cABE-2.0在靶腺嘌呤处实现了33.33%的A>G转换,而cABE-1.0为17.67%,传统ABE8e为13.67%(图6B)。
为了评估功能挽救,将编辑后的CG-4细胞分化为Mbp阳性的OLs,并对PLP亚细胞定位和形态参数进行量化。在PLPWT细胞中,PLP均匀分布在细胞体和突起中(体与突比例0.22:0.78),细胞表现出精细的分支(每个细胞15.01个分支;平均分支长度70.93μm)(图6C-F和补充图S8C)。相比之下,PLPA243V在核周积累(体与突比例0.69:0.32),分支严重减少(8.21个分支;长度31.96μm),重现了PMD表型。cABE-2.0纠正恢复了PLP向质膜和突起的分布(0.33:0.67),并部分挽救了形态缺陷(12.71个分支;66.79μm长度)(图6C-F)。
尽管cABE-2.0诱导了旁观者编辑,但在PLPA243V背景下,这些编辑在氨基酸水平上是沉默的(补充图S8D)。功能安全性在PLPWT细胞中得到进一步证实,未观察到显著的形态缺陷(体与突比例0.28:0.72;13.3个分支;67.16μm长度;图6C-F)。这些结果表明,cABE-2.0高效地纠正了致病性PLP1突变,恢复了蛋白质定位和OL形态,且未检测到有害效应。
cABE-2.0恢复突变OPCs的髓鞘功能
为了评估髓鞘形成潜力,将Flag标签的PLPWT或PLPA243VCG-4细胞与小鼠胚胎干细胞来源的神经元共培养(补充图S8E和F)。PLPWTOLs形成了广泛的Mbp阳性髓鞘,包裹着NF200阳性的轴突,93.3%的轴突显示出髓鞘共定位(图6G-J)。相比之下,PLPA243VOLs表现出严重的髓鞘形成障碍,仅有4.67%的轴突-髓鞘共定位。值得注意的是,用cABE-2.0纠正将髓鞘形成功能恢复到52.67%,比未纠正的突变OLs提高了11倍。这些发现表明,cABE-2.0高效地纠正了PLP1A243V突变,并挽救了少突胶质细胞分化和髓鞘形成能力。
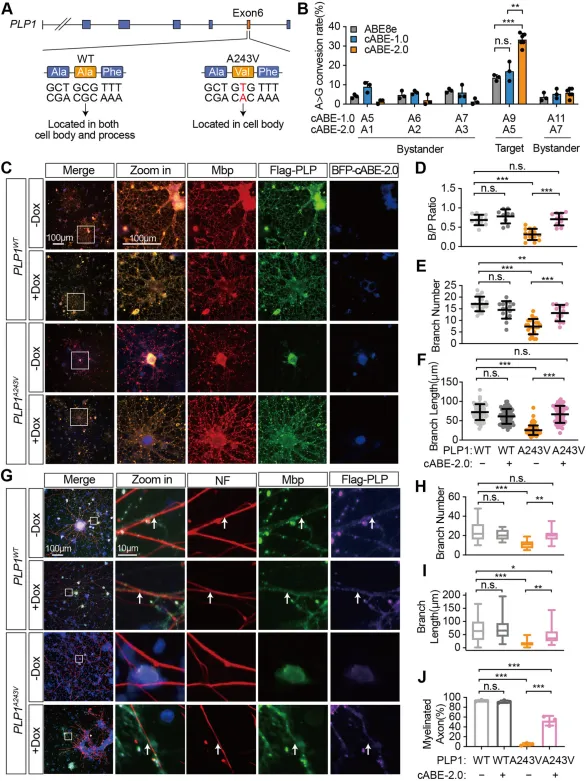
图6 |cABE-2.0纠正PLP1A243V突变并恢复少突胶质细胞分化。
(A) PLP1c.725C>T(p.Ala243Val,A243V)突变示意图。
(B) ABE8e和cABE变体在OLs中PLP1A243V突变位点的编辑效率(n=3个独立的生物学重复)。
(C) 编辑后的CG-4来源的OLs中Mbp(红色)、Flag-PLP(绿色)和BFP-cABE-2.0(蓝色)的免疫荧光图像。比例尺,100μm。
(D-F) 各组CG-4来源的OLs中PLP胞体与突起强度比(B/P比)(D)、分支数量(E)和分支长度(F)的量化。
对于(D)图,n=PLP1WT-Dox、PLP1WT+Dox、PLP1A243V-Dox、PLP1A243V+Dox组分别有14、11、10、9个独立细胞。
对于(E)图,n=相应组中分别有23、19、13、15个独立细胞。
对于(F)图,n=相应组中分别来自10、4、7、8个独立细胞的83、59、58、66个分支。
(G )CG-4来源的少突胶质细胞中Flag-PLP(紫色)、髓鞘碱性蛋白(Mbp,绿色)和神经丝蛋白(NF,红色)的免疫染色。箭头指示有髓鞘的轴突。
(H–J) 各组CG-4来源的少突胶质细胞中分支数量(H)、分支长度(I)和有髓鞘轴突比例(J)的量化。
对于(H)图,n=PLP1WT-Dox、PLP1WT+Dox、PLP1A243V-Dox、PLP1A243V+Dox组分别有23、22、23、23个独立细胞。
对于(I)图,n=相应组中分别来自5、5、7、6个独立细胞的92、94、77、76个分支。
对于(J)图,n=每组3个独立的显微镜视野。
(B,D-F,H-J)图中的数据以平均值±标准差表示。P值使用单因素方差分析(B,D-F,H-J)计算。*P<.05,**P<.01,***P<.001。
cABE-2.0在体外抑制PLP1表达
PLP1重复是PMD中最普遍的突变,约占病例的50%–70%(补充图S8A)。目前的疗法,包括反义寡核苷酸和RNA干扰,仅提供短暂的抑制,而不纠正潜在的基因组病变。通过腺嘌呤碱基编辑起始密码子(ATG→ACG)实现永久性敲低,提供了一种潜在的基因组解决方案。为此,用靶向PLP1起始密码子的ABE变体转导人MO3.13细胞和小鼠原代OPCs(补充图S9A和B)。在MO3.13细胞中,cABE-2.0实现了53.33%的编辑效率,显著高于eNme2-CABE8e(21.67%)、cABE-1.0(9.00%)或ABE8e(3.67%)。在小鼠OPCs中,cABE-2.0达到33.67%的效率,而eNme2-CABE8e为10.33%,其他变体<1%(补充图S9C)。
使用等位基因特异性定量实时PCR评估功能敲低,选择性扩增具有编辑起始密码子的转录本(补充图S9D和E)。在小鼠OPCs中,cABE-2.0将突变转录本减少了62.16%,与CRISPRi实现的49.70%相当,而eNme2-CABE8e减少了32.46%(补充图S9D和E)。蛋白质印迹证实,cABE-2.0将Plp蛋白表达降低至51.62%,优于ABE8e(96.98%)、cABE-1.0(82.50%)、eNme2-CABE8e(56.51%)和CRISPRi(54.86%)(补充图S9F和G)。这些结果表明,cABE-2.0在基因组和蛋白质水平上有效抑制了Plp1。
cABE-2.0能够在体内实现少突胶质细胞特异性的Plp1抑制
为了评估cABE-2.0是否能在体内抑制Plp1表达,我们使用Cre依赖性双floxed反向取向设计,将系统包装到AAV-PHP.eB载体中,以实现少突胶质细胞特异性表达。简而言之,我们将Cas9模块和TadA模块注射到8周龄Plp1-CreERT2小鼠的胼胝体中,随后给予他莫昔芬以诱导少突胶质细胞特异性的cABE-2.0重组。同时,接受表达eNme2-CABE8e或mCherry的AAV的小鼠作为对照。通过mCherry表达评估的皮质少突胶质细胞转导效率在各组间具有可比性(图7A)。
在他莫昔芬给药后第7天,对分选的mCherry阳性细胞进行Sanger测序显示,AAV-cABE-2.0实现了16.33%的编辑效率,显著高于AAV-N1组或AAV-mCherry组(图7B)。到第28天,免疫荧光分析表明,AAV-cABE-2.0处理组的Plp蛋白水平降至AAV-mCherry组的37.29%,而AAV-N1组为97.55%(图7C-E)。
为了在单细胞分辨率下评估Plp抑制,我们在径向距离20∼100μm的范围内,量化了单个mCherry阳性少突胶质细胞周围微环境中的平均荧光强度。虽然AAV-mCherry和AAV-N1处理未显示显著的Plp减少,但AAV-cABE-2.0处理导致了明显的Plp抑制,表明有效的敲低(图7F-G)。
鉴于Plp1缺陷会导致小鼠髓鞘形成不良和运动缺陷,我们使用转棒测试评估了功能后果。与AAV-N1组和AAV-mCherry组相比,接受AAV-cABE-2.0处理的小鼠表现出显著受损的运动表现。这些结果表明,cABE-2.0在体内有效地抑制了少突胶质细胞中的Plp1,为PLP1重复相关PMD的潜在治疗应用提供了原理验证。
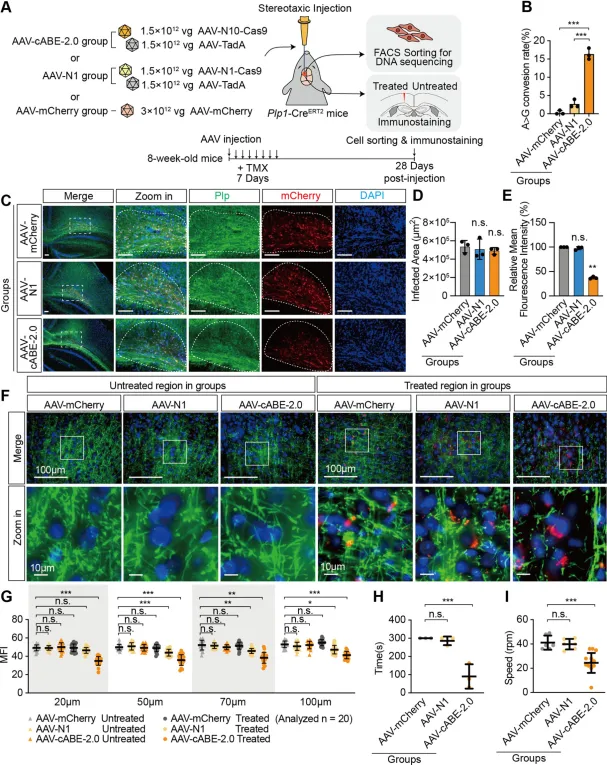
图7 |cABE-2.0通过ATG编辑在体内抑制Plp1。
(A) 将AAV-PHP.eB(3×10^12vg)立体定位注射到8周龄Plp1-CreERT2小鼠的实验时间线。注射后4周取脑进行FACS和测序。
(B) AAV-cABE-2.0、AAV-N1和AAV-mCherry组在mCherry阳性细胞中Plp1ATG位点的编辑效率(n=3个独立的生物学重复)。
(C) 显示mCherry感染区域和Plp表达的代表性图像。比例尺,100μm。
(D) AAV-mCherry、AAV-N1和AAV-cABE-2.0组中mCherry面积的量化(n=3个独立的生物学重复)。
(E) 相应组中mCherry阳性区域Plp平均荧光强度(MFI)的量化(n=3个独立的生物学重复)。
(F) 相应组中未处理(左)和处理(右)区域Plp(绿色)和mCherry(红色)的代表性免疫荧光。比例尺,100μm(合并),10μm(放大)。
(G) 相应组中,围绕mCherry阴性核周区(未处理)或mCherry阳性核周区(处理)的20、50、70和100μm范围内Plp荧光强度的量化(n=每组20个独立细胞,来自三个独立的生物学重复)。
(H,I) AAV处理小鼠的恒定速度转棒(H)和加速速度转棒(I)性能分析。数据显示为个体测试测量值及平均值±标准差。每组由三只生物学独立的小鼠组成。
在(H)图中,每只小鼠进行一次测试(每组n=3次测试)。
在(I)图中,每只小鼠进行多次测试,每组分别产生6、6和12次测试测量值。统计分析使用小鼠水平的平均值进行。
数据以平均值±标准差表示(B,D,E,G,H,I)。P值使用单因素方差分析(B,D,E,G,H,I)计算。*P<.05,**P<.01,***P<.001。
讨论
在本研究中,我们开发了cABEs,在保持紧凑、AAV兼容架构的同时,实现了增强的靶向DNA编辑和显著降低的RNA脱靶效应。优化后的cABE-2.0高效地纠正了与严重PMD相关的突变,恢复了少突胶质细胞的形态和髓鞘形成能力。这些结果确立了脱氨酶活性的空间集中,作为一种独特且有效的策略,用于改善在难以编辑的细胞类型(如少突胶质细胞)中的基因组编辑。
先前增强ABE性能的方法主要侧重于通过TadA蛋白质工程或编辑器过表达来增加脱氨酶催化活性或表达水平。虽然这些策略可以改善靶向编辑,但它们同时会提高全转录组范围的RNA脱靶活性,反映了活性和特异性之间的基本权衡,这限制了治疗应用。这种限制在少突胶质细胞中尤为突出,其中限制性的染色质环境减少了有效的靶点结合,突显了仅靠催化优化不足以克服”低修复-高脱靶”的限制。
我们的结果表明,脱氨酶模块的空间重组提供了一种替代解决方案。使用SunTag系统,cABE模块被主动转运到细胞核,并形成具有与LLPS一致特性的动态斑点。这些凝聚物将TadA*集中在基因组靶点,增强了局部催化效率,同时限制了可用于RNA底物的自由扩散酶。值得注意的是,编辑效率和斑点形成与SunTag价态呈非单调关系,在N端10×GCN4重复处达到峰值,并在更高拷贝数时下降,这与先前的研究一致,表明过度的多价性会降低分子流动性和凝聚物流动性。这一观察结果强调,最佳的空间组织,而非最大的多价性或催化活性,对于高效和特异的碱基编辑至关重要。
虽然cABE-2.0实现了高编辑效率和特异性,但相邻的旁观者腺嘌呤仍可能被修饰,这与TadA介导的脱氨固有的序列窗口一致。通过优化的gRNA设计或工程化的脱氨酶变体来解决这一限制,代表了未来改进的一个合理方向。
总的来说,这些发现确立了通过类LLPS组装实现的空间集中,作为精确基因组编辑的一个可推广的原理,补充了基于蛋白质工程的现有策略。cABE平台在多样化的人类和神经细胞类型中表现出稳健的性能,高效地纠正致病性突变,并在体外和体内实现少突胶质细胞特异性的基因调控,为PMD和其他髓鞘疾病的潜在治疗干预提供了机制和技术框架。
本文为非医疗专业人士自愿翻译,仅供患者家属学习参考。如果文中有歧义,请按题目(文首截图)自行查询英文原文核准。
文献翻译丨嘉鸿妈妈
文档整理丨琛宝爸爸
审核校对丨希希妈妈

微信号丨Sameen_33
公众号丨佩梅病之家
求点赞

求分享

求喜欢
