 夜雨聆风
夜雨聆风





从GEO下载转录组数据:差异基因、火山图、热图、GO/KEGG功能富集分析方法

如何成为专业的生信人,那就是搞到质量高得数据。在数据库中,上传者往往不会将自己整理好的数据直接上传,而是放一个测序的原始数据,那我们下载下来是无法直接使用的,需要进行专业处理后得到基因表达矩阵。今天分享一个GEO数据库上载录的于2022年发表的人的RNA-seq测序数据,并教大家如何下载和预处理(将原格式转换为基因表达矩阵)
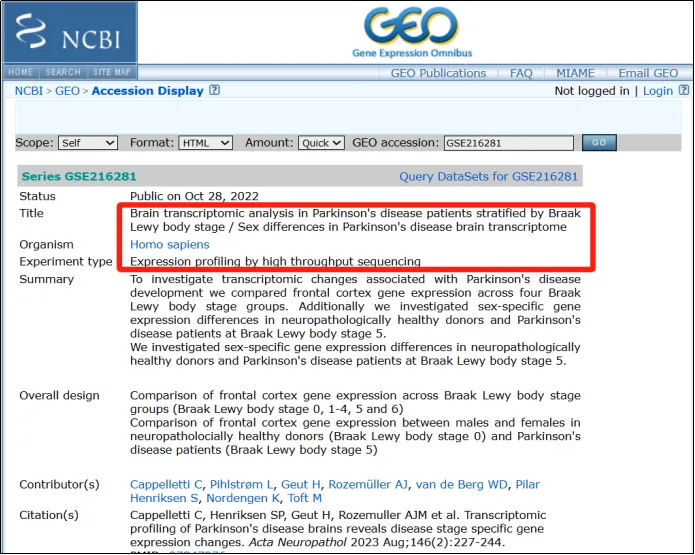
演示数据获取网址如下:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE216281
(1)可以发现作者这里仅提供了一个文件,是txt文件,算是比较友好的。点击http下载。
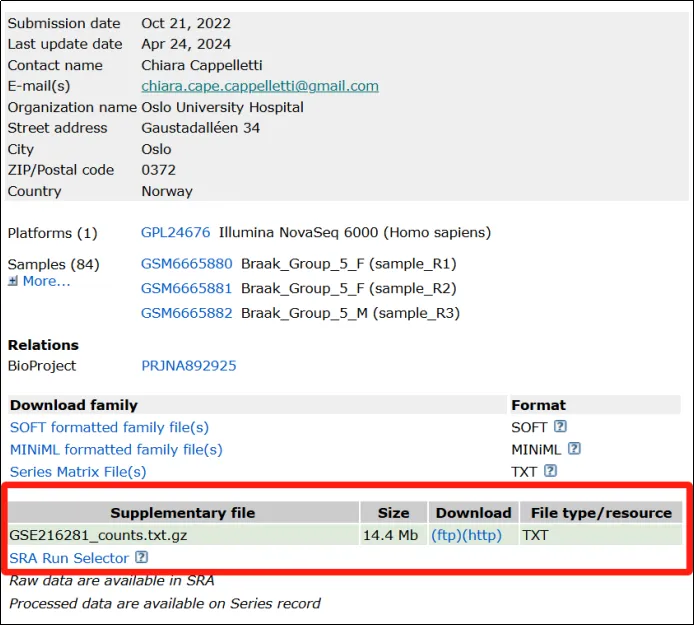
小贴士:在GEO 数据库的下载栏中,FTP 和HTTP 是两种用于下载数据的协议。
1. FTP(File Transfer Protocol):通过FTP 下载数据时,会提供一个 FTP 地址,用户可以通过工具连接到 GEO 的服务器并下载文件。适合大文件或批量下载。需要使用支持 FTP 的客户端(如 FileZilla)或命令行工具(如 wget)进行下载。
2. HTTP(Hypertext Transfer Protocol):点击链接即可在浏览器中直接下载文件。
(2)解压缩下载的文件
(3)用excel打开txt文件
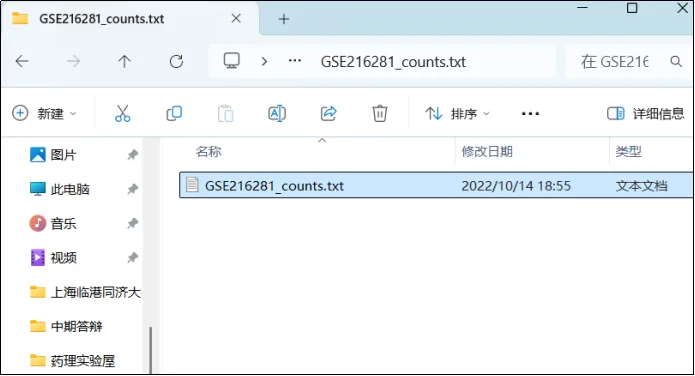
(4)观察这个文件,A1单元格为所有样本名称
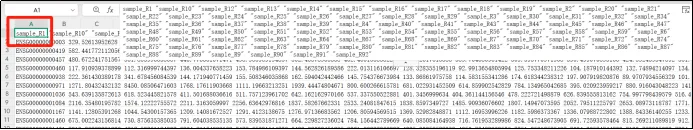
(5)A2单元格开始为基因探针和每个样本的该基因FPKM值
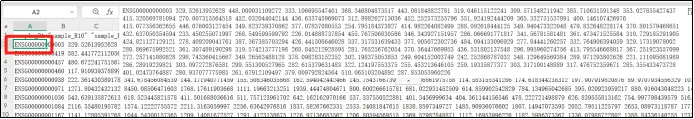
(6)我们需要做的就是,将A1中每个样本独立占据单元格,依次按B1、C1等分开;A2仅为基因探针,基因表达值依次占据B2、C2等单元格。如图所示,并且下面将演示如何操作。
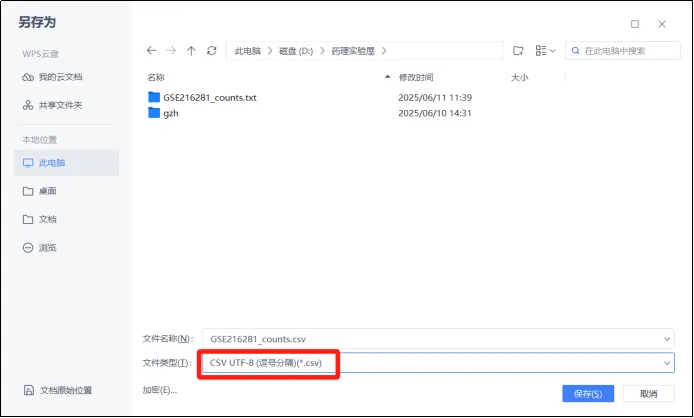
(7)首先将文件另存为csv文件。
(8)用R语言处理该csv文件,运行代码即可。注意R操作路径为该文件所在位置(代码为药理实验屋团队编写,点赞 + 爱心,评论区留言即可领取)。
(9)打开代码运行后保存的文件,可以看到样本名称已经分开,并依次填充到后面单元格,与基因表达值一一对应;基因探针为第一列。现在只需要手动删除第一行,并将A2单元格NA改成Gene Symbol。
(10)现在使用R将基因探针对应成基因名称(代码为药理实验屋团队编写,点赞 + 爱心,评论区留言即可领取)。
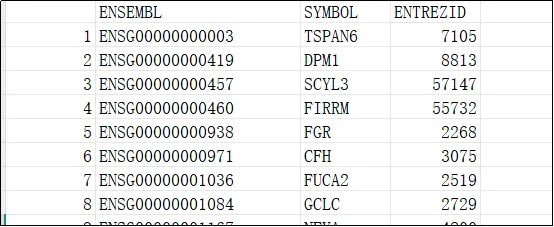
(11)打开保存的文件,可以发现基因名称与探针一一对应。
(12)下面将上面三栏内容复制并插入表达矩阵中,并用函数索引进行基因的填充替换。黄色为原基因探针,橙色为复制内容,GENE栏为将要填充的基因探针对应的基因。
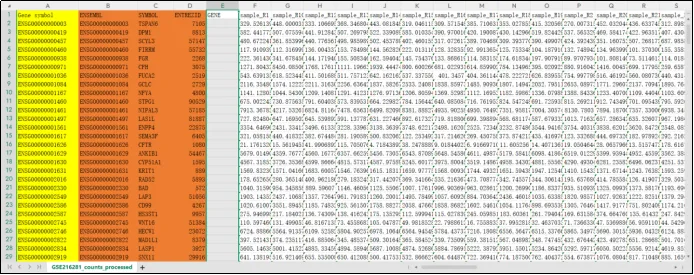
(13)使用VLOOKUP函数,公式:=VLOOKUP(@A:A,B:D,2,FALSE)。
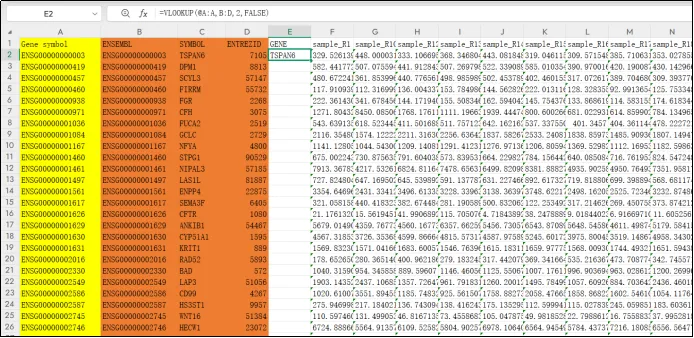
(14)选中单元格,CTRL+D进行填充,补全全部基因。
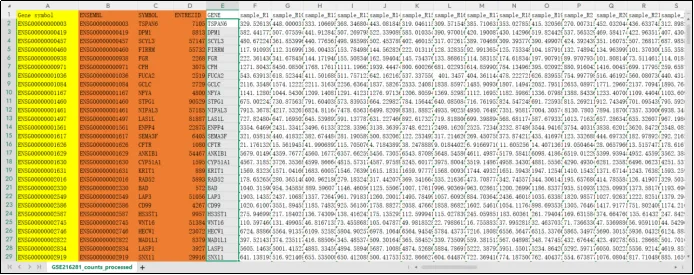
(15)将GENE这一栏复制为仅数值插入右侧。
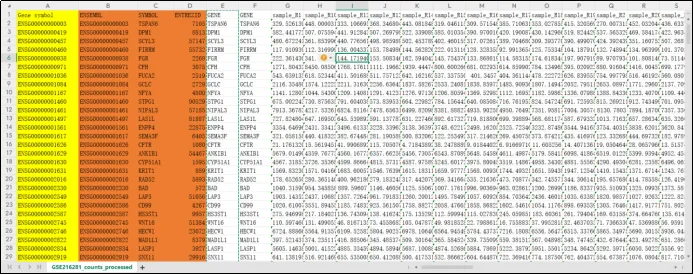
(16)删除前5列即可得到完整的基因表达矩阵。
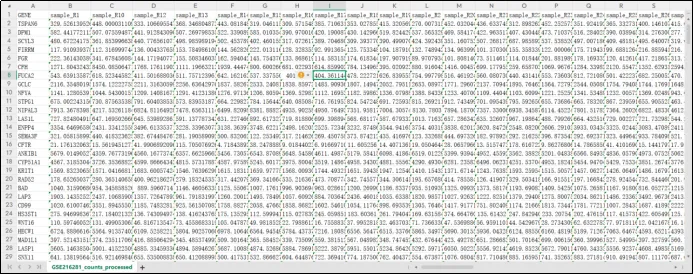
(17)将GENE按升序排列。
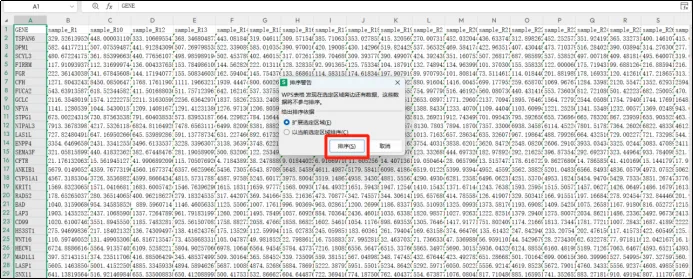
(18)下拉到基因名为N/A的地方并删除。出现基因为N/A的原因是由于基因探针与GENE名称的对应关系在不断变化,或者每个测序公司有非公用探针。这种时候只能忽略这些值,或者更新自己的R包,或者联系文章作者。
(19)最后,将样本名更改成正常和疾病。回到GEO数据库中查看每个样本代表的是正常还是疾病。可以发现Braak_Group_0是正常,其余均代表发病,只是病情阶段不同。

(20)在基因表达矩阵中更改样本信息。
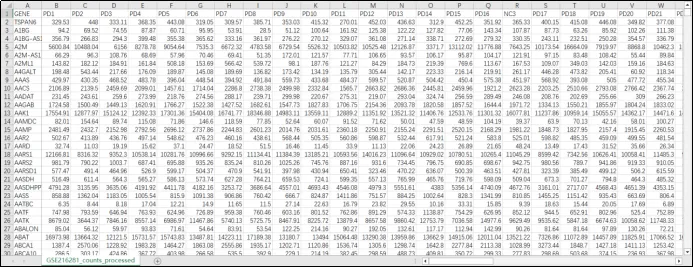
(21)新建一个CSV文件,将样本信息归组,用于后续差异基因分析用。
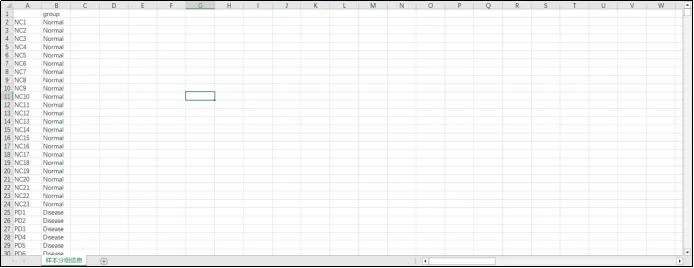
(22)读取基因表达矩阵,合并相同基因行,重新保存文件。因为一个基因会对应多个探针,所以转换ID会产生相同的基因,这时需要进行合并。
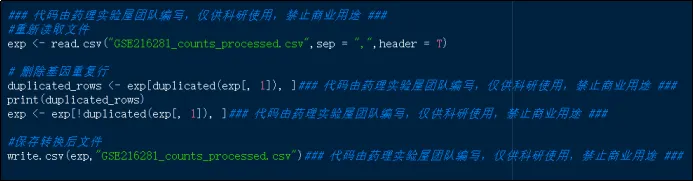
(23)重新读取基因表达矩阵,处理数据,包括取对数、均一化、去除低表达等。
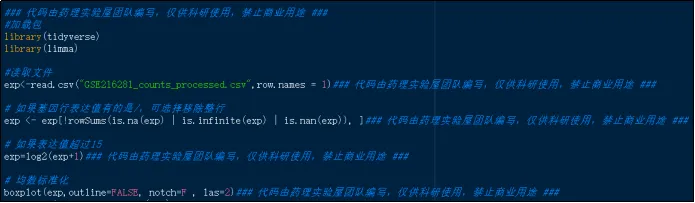
(24)均一化展示图。
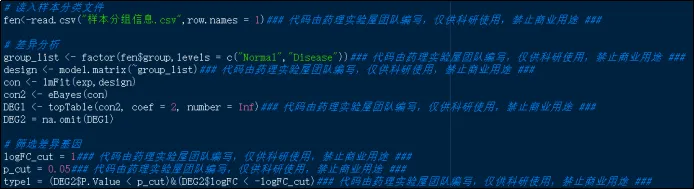
(25)读入样本分组信息,以logFC 为 1、 P.Value 为 0.05 为筛选目标进行差异基因分析。
(26)保存后的差异基因中注明了每个基因是上调还是下调。
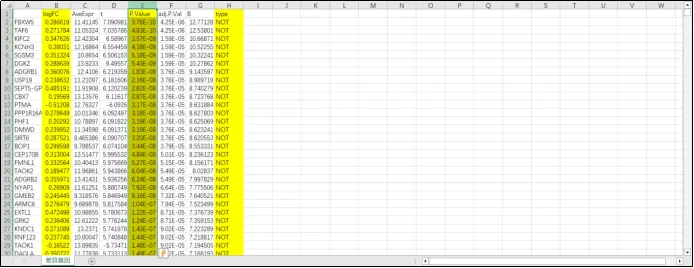
(27)利用差异基因绘制火山图。
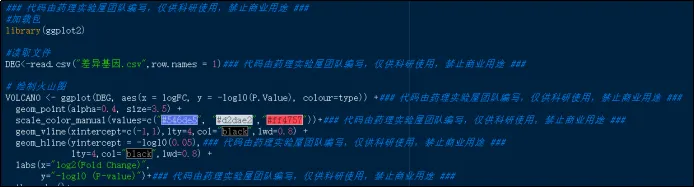
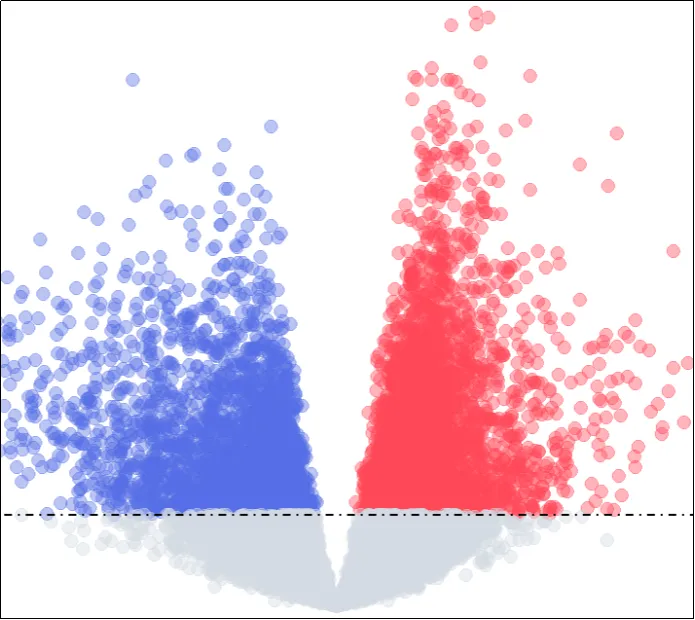
(28)火山图展示图。
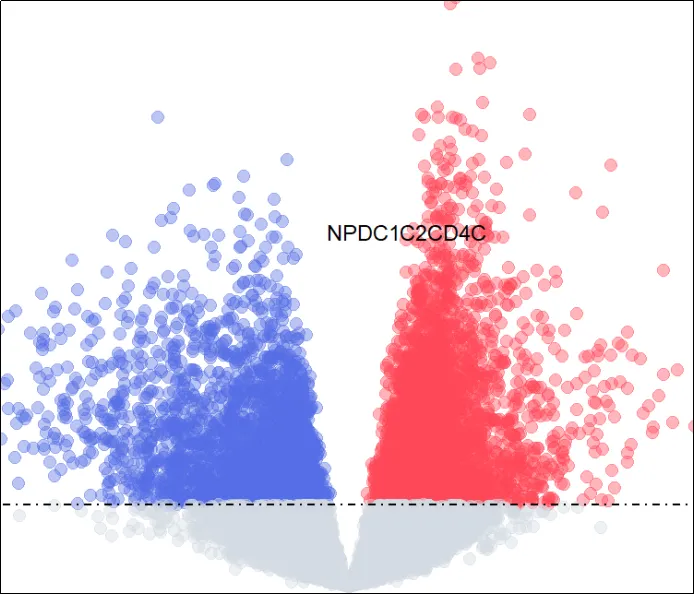
(29)添加基因。
(30)读入基因表达矩阵,绘制热图。
(31)热图展示图。
(32)换个聚类算法。

(33)换个颜色。
(34)利用差异基因进行富集分析。可以使用上调、下调、上调+下调或自定义基因进行分析。将需要分析的基因保存为CSV文件。
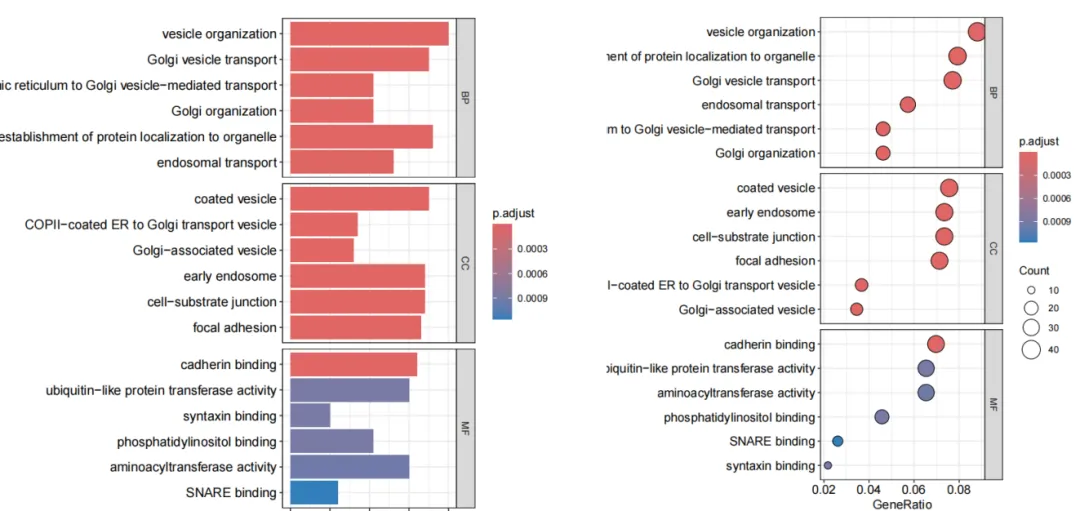
(35)GO/KEGG功能富集分析展示图。
该代码通过网盘分享。点赞👍加爱心❤,评论区留言领取~

