 夜雨聆风
夜雨聆风





分子对接软件哪个更好用?
做分子对接时,很多人最先问的是:哪个软件更好用?
但如果真要把这个问题讲清楚,不能只是简单列一个软件名单。因为“分子对接”本身并不是完全统一的问题,至少可以分成三大类:蛋白–小分子对接、蛋白–蛋白对接,以及蛋白–DNA/RNA 对接等。这三类问题在搜索空间、打分逻辑、柔性处理和结果解释上都有明显差别,所以主流软件也自然分成了不同路线。
如果把这些软件放在一起看,可以先做一个大致分类:做蛋白–小分子时,大家最常接触的是 AutoDock 系列、Schrödinger、MOE、Discovery Studio等;做蛋白–蛋白时,更常见的是 HADDOCK、HDOCK、ZDOCK、ClusPro、GRAMM、RosettaDock等;到了蛋白–DNA/RNA,值得关注 HDOCK、NPDock、3dRPC、P3DOCK,以及 AlphaFold 3等工具。
重要的并不是把软件背下来,而是要知道:不同工具到底擅长解决什么问题。下面就按这三类体系,把主流路线分别梳理清楚。
一、蛋白–小分子对接软件:最成熟、应用最广的一类
蛋白–小分子对接是目前应用最广的一类分子对接。它的核心任务通常是预测:小分子会结合在蛋白的什么位置、以什么姿态结合、可能涉及哪些关键残基,以及不同化合物的大致排序。因为应用面最广,所以这一类软件的发展也最成熟,既有经典开源软件,也有功能非常完整的商业平台。
1)AutoDock4:经典老牌软件,优势在于基础扎实、可控性强
AutoDock4 是 AutoDock 体系里最经典的一代。AutoDock4 实际上由 AutoDock和 AutoGrid两部分组成:AutoGrid 先为受体预计算网格图,AutoDock 再在这些网格基础上完成配体搜索和打分。也正因为这种设计,AD4 的流程相对清楚,很多参数和步骤都比较“可解释”,所以它长期被广泛用于教学、方法学研究以及传统蛋白–小分子对接工作。
从实际使用感受来说,AutoDock4 的优势不只是“经典”。它更适合那些希望对搜索参数、网格设置、局部环境定义有更多控制的人。很多研究者现在仍然喜欢用它做复现实验、方法比较,或者处理一些标准化程度较高的蛋白–配体体系。它的文献积累也非常深,很多早期和中期的 docking 文章都是基于 AD4 完成的。
2)AutoDock Vina:开源体系里最普及的通用工具之一
如果说 AD4 更像经典路线,那 AutoDock Vina更像现在最常见的开源起点。Vina 官方文档把它定义为“最快、使用最广的开源 docking engine 之一”,并强调它是一个相对 turnkey 的对接程序,基于简单打分函数和快速梯度优化搜索。Vina 之所以被广泛接受,很重要的原因是它比较好地兼顾了速度、易用性和构象搜索能力。
Vina 的实际优势主要体现在几个方面:第一,它上手门槛相对较低,很适合常规蛋白–小分子 docking 和中等规模虚拟筛选;第二,它运行速度快,适合做批量任务;第三,它依托 AutoDock 社区,教程多、经验多、可复现性也相对容易建立。对很多课题组来说,Vina 往往是从“能把 docking 跑起来”到“能系统做项目”之间最自然的一站。
3)AutoDockFR(ADFR):以受体柔性为核心,同时支持特定类型的共价对接
AutoDockFR(ADFR)是 AutoDock 体系中比较有代表性的柔性受体对接方案。它的核心特点,是允许用户预先指定受体中的一部分侧链作为柔性区域参与搜索,而不是把整个受体完全视为刚体。ADFR 的原始论文强调的也是这一点:它主要解决的是蛋白–配体对接中受体侧链柔性与诱导契合的问题。
这类方法的优势很明确:当配体进入结合口袋时,如果关键残基侧链需要发生重排,ADFR 相比纯刚体受体方法,往往更容易得到贴合更合理的结合构象。因此,它特别适用于口袋附近存在明显侧链调整、局部诱导契合效应较强的体系。对于很多“普通 Vina 能跑,但总觉得口袋贴合不够自然”的项目,ADFR 往往是一条很有价值的补充路线。
另外,ADFR 配套的 AGFR/ADFR 工作流也支持共价对接。Scripps 官方教程专门给出了 covalent docking 的示例,说明这套工具链可以用于已知成键位点、已知共价连接关系前提下的共价配体建模与再对接。也就是说,ADFR 不只是能做柔性受体 docking,在特定场景下也可以处理共价配体问题。
不过从方法定位上说,ADFR 的第一标签仍然是柔性受体 docking;共价对接更适合理解为它的重要扩展能力,而不是唯一或最核心的功能。换句话说,ADFR 最值得强调的优势仍然是:既能处理口袋侧链重排,又能在特定设定下兼顾共价结合场景。
4)AutoDock CrankPep(ADCP):AutoDock 体系中专门面向肽–蛋白对接的路线
如果说 AutoDock4、Vina 和 AutoDockFR 主要还是围绕蛋白–小分子 docking 展开,那么 AutoDock CrankPep(ADCP)则是 AutoDock 体系里非常值得单独拿出来讲的一条线,因为它的定位不是普通小分子,而是 peptide docking。Scripps 官方页面明确写到,ADCP 是一种专门用于肽对接的 AutoDock 引擎,它把蛋白折叠领域的方法与刚性受体的亲和力网格表示结合起来,在受体形成的能量景观中对肽进行折叠,并同时优化肽与受体之间的相互作用;其核心搜索方式是 Monte Carlo。官方还提到,ADCP 可以直接以肽序列字符串作为输入,并已成功应用于长度约 20 个氨基酸以内的肽。
ADCP 的优势很明显。普通小分子 docking 软件面对肽时,往往会遇到一个很现实的问题:肽的主链和侧链自由度明显更高,不能简单当成“稍微大一点的小分子”处理。而 ADCP 的思路,正是把“肽会折叠、会变构象”直接纳入 docking 过程。2019 年的论文把它概括为一种“combining folding and docking”的方法,用于预测 protein–peptide complexes。也正因为这样,ADCP 特别适合用于短肽–蛋白、柔性肽–蛋白这类体系。
另外,ADCP 后续还扩展到了环肽 docking。相关研究明确指出,新版本的 AutoDock CrankPep 可以处理通过 backbone 和/或 side chain cyclization 形成的 cyclic peptides。AutoDock 系列中专门解决肽类配体,尤其是高柔性肽和部分环肽问题的重要工具。
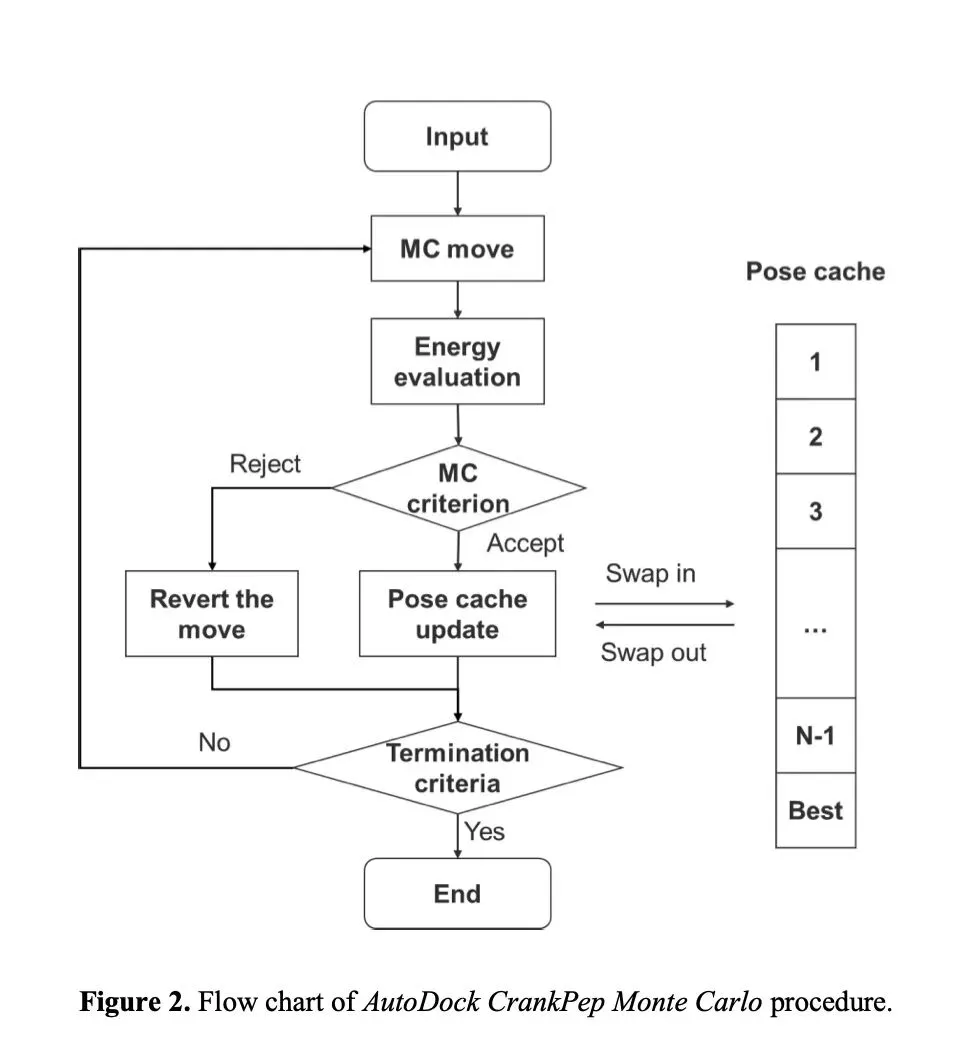
5)Schrödinger / Glide:商业平台里非常成熟的一条线
在商业软件里,Schrödinger基本是绕不开的平台。严格来说,Schrödinger 不是单独一个“对接软件”,而是一整套结构基础药物设计平台,其中最核心的蛋白–小分子 docking 模块通常就是 Glide。官方产品页把 Glide 定位为准确、通用的 ligand–receptor docking 程序,可用于虚拟筛选、结合模式预测以及交互式三维设计。官方白皮书还进一步说明,Glide 在排序和构象筛选中会结合 GlideScore 与 Emodel 等不同评分思路。
Glide 的优势,很多时候不只是“对接精度高”这一句话能概括的。它真正强的地方在于工作流完整:前面可以接蛋白准备、网格生成、口袋定义,后面可以自然接上相互作用分析、重打分,甚至与更深入的自由能和药化设计模块衔接。也正因此,Glide 更像是一个成熟工业化流程中的核心节点,而不仅仅是一段单独的 docking 程序。
6)MOE:一体化分子建模平台,适合把前处理、对接和后分析放在一起
MOE(Molecular Operating Environment)也是分子对接领域很常见的一体化平台。Chemical Computing Group 官方产品页把它描述为一个集成式分子设计平台,覆盖活性位点分析、蛋白–配体相互作用分析、诱导契合对接、片段设计等功能。MOE 资料还特别提到,它提供多种 docking 方式,包括 induced-fit docking、template docking 和 covalent docking。
MOE 平台的实际优势,在于它的“整合感”很强。用户可以在同一个环境中完成口袋识别、配体准备、对接、相互作用可视化以及一定程度的后处理。对很多希望流程集中、界面统一、分析顺手的用户来说,MOE 的吸引力就在这里。它并不只是一个“跑 docking 分数”的工具,而是更偏向综合建模与分析平台。
7)Discovery Studio(DS):平台型路线,内部包含多种对接模块
Discovery Studio常被很多人简称为“DS 对接”,但严格来说,DS 不是单一算法,而是一个整合式生命科学建模平台。BIOVIA 官方概览里写得很明确:Discovery Studio 集成了多种计算方法,其中蛋白–配体对接相关模块包括 GOLD、LibDock、CDOCKER等,而蛋白–蛋白对接则集成了 ZDOCK。
如果单说蛋白–小分子这条线,DS 里比较常被提到的是 LibDock和 CDOCKER。LibDock 更偏向基于口袋特征点进行快速筛选,适合高通量初筛;CDOCKER 则是基于 CHARMm 力场的一类对接方案,更偏向构象优化和精细打分。再加上 DS 自身在可视化和后续处理方面的整合能力,它在很多项目里更像一个平台化建模环境,而不是某一个单点算法。
二、蛋白–蛋白对接软件:关注的核心是界面,而不只是口袋
和蛋白–小分子不同,蛋白–蛋白对接的难点通常不在于把一个小分子塞进明确口袋,而在于预测两大片表面怎样形成合理界面。也正因为这样,这类软件更强调的往往不是传统意义上的“口袋匹配”,而是界面互补、刚体或半柔性搜索、聚类分析,以及实验信息约束。目前比较主流的对接软件程序包括 HADDOCK、HDOCK、ZDOCK、ClusPro、GRAMM、RosettaDock等,同时也有一些更偏 AI 复合物预测的路线逐渐进入视野。
1)HADDOCK:信息驱动型蛋白–蛋白对接的代表
HADDOCK 的全名是 High Ambiguity Driven biomolecular DOCKing。从最早的经典论文到 Bonvin 实验室的软件页面,它的定位都非常明确:这是一个基于生化或生物物理信息来驱动 docking 的平台。也就是说,如果你已经有突变位点、NMR 化学位移扰动、交联、已知界面残基等先验信息,HADDOCK 可以把这些信息作为限制条件纳入对接。
HADDOCK 的核心优势就在这里:它并不只是盲目搜索所有可能构象,而是允许用户把实验知识真正整合进建模流程。因此在很多真实课题里,尤其是已经有一定实验线索的蛋白–蛋白体系,HADDOCK 往往很受欢迎。它不仅能做蛋白–蛋白,也覆盖更广义的 biomolecular docking。
2)HDOCK:模板与自由对接结合,适合快速获得初始复合物
HDOCK 官方帮助页面和对应文章都指出,这个服务器支持蛋白–蛋白以及蛋白–DNA/RNA 对接,并采用 template-based 与 template-free 相结合的 hybrid docking strategy。它的一个很明显的优势是:即便用户手里只有序列或结构信息,也有机会快速获得一个比较完整的复合物预测流程。
从实际使用角度说,HDOCK 的吸引力主要在于上手快、界面友好、适用对象广。对于很多想先拿到一个蛋白–蛋白初步复合物,或者同时还涉及核酸复合物的项目来说,HDOCK 往往是很实用的入口平台。它不是强调特别复杂的交互设置,而是强调“快速给出一个可用的初始模型”。
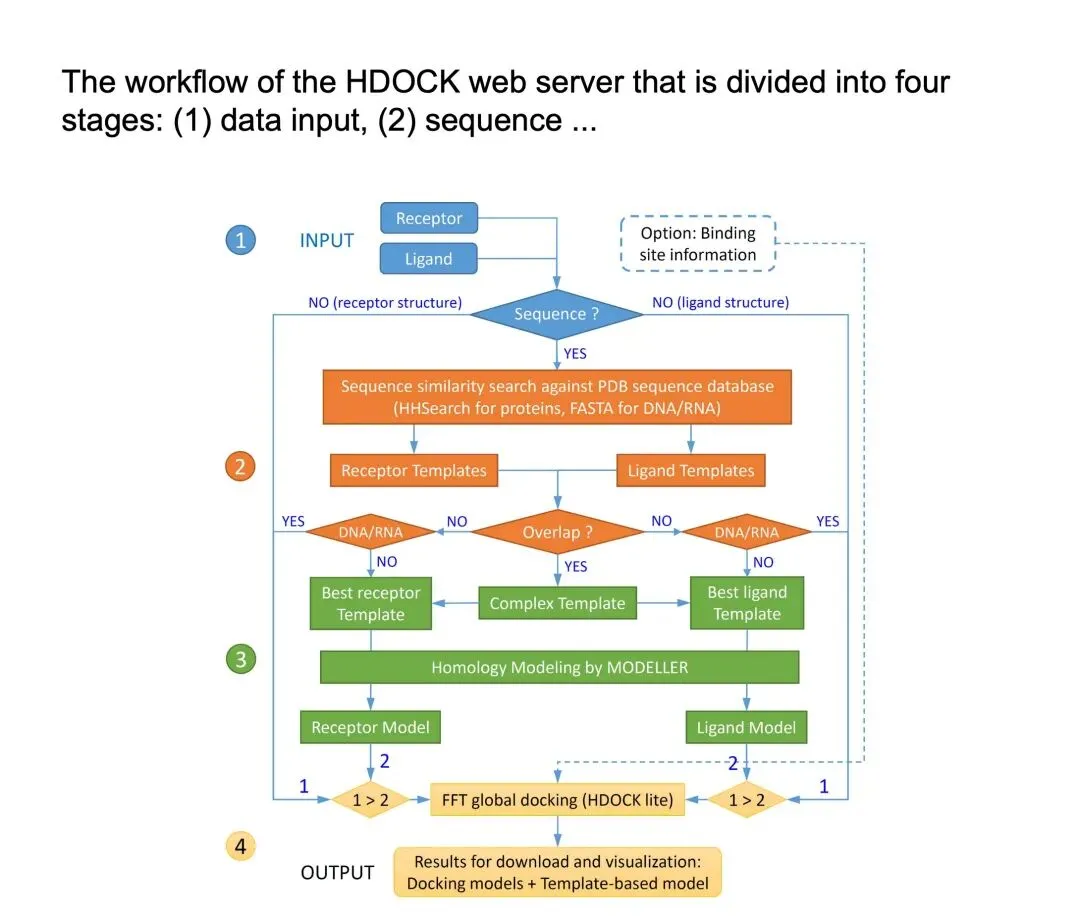
3)ZDOCK:经典刚体蛋白–蛋白对接程序
ZDOCK是蛋白–蛋白 docking 领域的经典名字之一。虽然很多用户现在更多是在 Discovery Studio 等平台里接触到它,但这恰恰说明了它在商业和应用平台中的核心地位。ZDOCK 的传统优势主要在于刚体搜索成熟、采样效率高、适合作为蛋白–蛋白初始构象生成工具。
很多项目里,ZDOCK 的价值在于先生成一批候选界面,再结合聚类、界面分析和后续精修来筛选。也因此,它很适合用来做结构域之间、已知互作蛋白之间的初始界面探索。在不少平台里,ZDOCK 实际上承担的是“第一轮全局对接”的角色。
4)ClusPro:以聚类思想著称的蛋白–蛋白对接服务器
ClusPro是蛋白–蛋白 docking 里很有代表性的 web server。它的帮助页非常直白:平台提供 balanced、电荷偏好、疏水偏好、抗体模式等不同选项,并强调“好的结果往往需要和实验知识一起使用”。这反映出 ClusPro 的一个鲜明特点:它很强调大量采样后的聚类筛选。
对很多用户来说,ClusPro 的优势在于它非常适合做自动化的蛋白–蛋白初筛。你不一定一开始就知道准确界面,但它能给你多个不同物理倾向下的代表性聚类结果,帮助你从整体上判断复合物可能落在哪一类界面上。它特别适合作为“先看整体界面格局”的工具。
5)GRAMM / GRAMM-X:经典 FFT 路线,强调全局能量景观搜索
GRAMM是老牌蛋白–蛋白 docking 路线,其名称就来自 Global RAnge Molecular Matching。GRAMM 通过系统性映射分子间能量景观,来预测对应稳定和瞬态相互作用的一系列 docking poses。GRAMM-X 则是在原始 FFT 思路基础上,结合平滑势能、精修和知识型打分而发展出来的公共服务器。
GRAMM 类方法的优势在于,它对全局搜索和界面景观描述比较有代表性,适合用来快速扫描可能的相互作用方式。对于那些更看重“可能有哪些界面解”,而不是一开始就只盯着一个最优答案的项目,GRAMM 仍然有自己的位置。
6)RosettaDock:更强调界面精修、柔性优化和多尺度建模的一条路线
如果说 ZDOCK、ClusPro、GRAMM 这类工具更偏向于先做全局采样、先找可能的界面构型,那么 RosettaDock更值得被理解为一条更强调界面精修和局部优化的路线。Rosetta 官方文档把 RosettaDock 描述为一种 Monte Carlo based multi-scale docking algorithm:先在低分辨率阶段进行粗粒化搜索,再进入高分辨率全原子 refinement 阶段,同时优化复合物的刚体取向和界面侧链构象。也就是说,它并不只是“把两个蛋白摆在一起试一试”,而是会进一步处理界面附近残基在结合过程中的重排和适配问题。
这也是 RosettaDock 和很多经典刚体 docking 程序相比,一个很重要的区别。蛋白–蛋白结合界面并不总是完全刚性的,很多体系在真正结合时,界面侧链会发生重新取向,局部主链也可能出现一定程度的适配。对于这类问题,只依赖刚体搜索往往不够,而 RosettaDock 的优势就在于:它不仅关注“能不能对上”,还更关注“对上以后,界面是否能被优化得更合理”。换句话说,它更适合处理那些初始界面已经大致有方向,但还需要进一步 refinement的场景。
从实际应用角度看,RosettaDock 往往特别适合以下几类问题。第一,已经通过实验、模板或其他 docking 工具拿到一个初始复合物候选模型,希望进一步优化界面细节;第二,体系中界面侧链柔性比较明显,希望在 docking 过程中兼顾一定的构象调整;第三,不只是想得到“一个可能能结合的姿态”,而是希望进一步比较不同界面构型在精修后的合理性。
因此,RosettaDock 在很多项目里的定位,并不是最前端的“快速初筛工具”,而更像是中后段的精修工具。它可以和其他软件形成很好的互补:比如先用 ZDOCK、ClusPro 或 HDOCK 做全局搜索,拿到若干候选复合物,再用 RosettaDock 对候选界面做进一步优化和筛选。这样理解会更准确。
另外,Rosetta 整个软件套件本身覆盖的并不只是单一 docking,而是更广义的大分子结构建模与设计框架。所以 RosettaDock 的优势,也不只是“有一个 docking 分数”,而在于它背后依托的是一整套更强调结构优化、能量评估和界面设计的建模思路。也正因为这样,RosettaDock 更适合那些愿意投入更多建模细节和计算资源,来换取更高界面优化自由度的课题,而不是追求最快速、最省事的一键式服务器结果。
如果用一句话概括,RosettaDock 最值得强调的特点不是“它也能做蛋白–蛋白 docking”,而是:它在蛋白–蛋白对接里,更偏向从候选构象走向界面精修,是连接‘初始对接结果’和‘更细化结构模型’之间的一条重要路线。
7)AlphaFold 3:不是传统 docking 软件,但已经进入复合物结构预测视野
AlphaFold 3严格来说并不是传统意义上的 docking 程序,但它已经显著改变了大家对复合物结构预测的讨论方式。2024 年 Nature 论文明确写到,AF3 采用更新后的 diffusion-based 架构,能够联合预测蛋白、核酸、小分子、离子和修饰残基等复合体系的结构与相互作用。也就是说,它不再局限于单个蛋白结构,而是开始覆盖多分子复合物。
AF3 的优势在于,它把“复合物结构预测”推进到了更高层次,尤其在蛋白–蛋白、蛋白–核酸甚至蛋白–小分子问题上都引发了大量关注。不过它的定位仍然更接近复杂体系结构预测模型,而不是传统 docking 中那种可控的局部参数搜索工具。所以在实际工作里,AF3 更适合作为一种强有力的新型预测路线,而不是简单替代所有经典 docking 软件。
三、蛋白–DNA/RNA 对接软件:核酸体系更强调界面特征和专门评分
蛋白–DNA/RNA 对接相比蛋白–蛋白更特殊。因为核酸带有明显的骨架电性、碱基堆叠和二级结构特征,所以很多普通蛋白–蛋白工具虽然也能跑,但不一定最能抓住核酸界面的特点。也正因此,除了 HDOCK 这种兼顾蛋白与核酸的大分子服务器外,NPDock、3dRPC、P3DOCK等专门面向蛋白–核酸复合物的工具也很重要。近年的综述也把 protein–nucleic acid docking和 binding affinity prediction单独作为一类问题来讨论。
1)HDOCK:蛋白–DNA/RNA 的通用入口之一
前面讲蛋白–蛋白时已经提到,HDOCK 官方明确支持 protein–DNA/RNA docking。它的优势之一,正是在于一套服务器同时覆盖蛋白–蛋白和蛋白–核酸,并且支持序列和结构输入。这使得 HDOCK 在实际课题里非常方便,尤其适合那些既做蛋白互作、又做蛋白–DNA 或蛋白–RNA 的团队。
对于核酸体系来说,HDOCK 的价值主要在于它给了一个比较稳妥的通用入口:如果你想先看一个蛋白和 DNA/RNA 的整体结合格局,HDOCK 往往是很实用的起点。不过它在这一类问题中的意义,更偏向于通用性强、上手方便、统一平台,而不是核酸专用评分本身。也就是说,HDOCK 在蛋白–核酸方向很实用,但它的特色并不在“核酸专门化”,而在“一套工具覆盖更广”。
2)NPDock:专门做蛋白–核酸复合物建模的服务器
NPDock的定位非常明确,它是一个专门用于 RNA–protein 和 DNA–protein complex structures建模的 web server。官方说明写到,它把 GRAMM 的全局大分子对接、统计势打分、聚类和局部精修结合在一起。也就是说,NPDock 不是简单把蛋白–蛋白方法生搬过来,而是专门围绕蛋白–核酸复合物建模设计了一整套流程。
NPDock 的优势主要在于两点:第一,它面向核酸体系本身;第二,它把“全局搜索—统计打分—聚类—精修”串成了一个比较完整的管线。对于很多需要快速建立蛋白–DNA/RNA 复合物初始模型的项目来说,这类平台很有实际价值。
3)3dRPC:更强调 RNA–蛋白界面特征的专门方法
3dRPC是一个更偏 RNA–protein的专用平台。相关论文指出,3dRPC 用于 3D RNA–protein complex structure prediction,其核心包括 RPDOCK 对接算法与针对 RNA–protein 界面的评分函数。换句话说,3dRPC 不是只做一般大分子碰撞,而是试图更细致地考虑 RNA–protein 接口的几何和相互作用特征。
因此,3dRPC 的一个突出优势就是它对 RNA–蛋白问题更“专门化”。如果研究重点就在 RNA–protein 识别,而不是泛化的大分子 docking,3dRPC 这类方法会显得更有针对性。
4)P3DOCK:模板与自由对接结合的蛋白–RNA 路线
P3DOCK的官方帮助页说明得很清楚:它是一个基于模板与自由对接相结合的protein–RNA docking server,其中自由对接部分采用 3dRPC,模板部分则走另一条结构相似性路线。也就是说,P3DOCK 的优势在于把模板信息与 de novo docking 思路放在同一平台中结合使用。
这类平台的实际价值在于,当体系存在可借鉴模板时,它能利用模板提高预测效率;当模板不足时,又还能回退到自由 docking。对于蛋白–RNA 体系来说,这种 hybrid 思路是非常有吸引力的。
5)AlphaFold 3:也正在进入蛋白–DNA/RNA 复合物预测主战场
AF3 的一个特别值得关注的地方,就是它明确把 DNA、RNA、小分子等都纳入联合建模范围。2024 年 Nature 论文已经说明,它能够预测包括 nucleic acids 在内的多组分复合物;后续综述和评估工作也都把 AF3 放到了蛋白–核酸复合物预测的重要位置上。甚至在后续一些评估中,AF3 在 protein–RNA复合物测试里表现优于多种传统 server,但也并非所有案例都完美。
因此,在蛋白–DNA/RNA 这一类问题里,AF3 的意义不只是“它也能做”,而是它已经开始明显改变大家对蛋白–核酸复合物预测的预期。不过和前面一样,AF3 更适合作为一种强有力的新型复合物结构预测路线来看待,而不是把它直接等同于传统 docking 软件。尤其在很多仍然需要局部可控搜索、参数设定和结果可解释性的场景里,经典 docking 工具依然有自己的位置。
四、这些软件到底该怎么选:不是谁最有名,而是谁更适合你的问题
把上面这些软件放在一起看,真正需要回答的其实不是“谁更强”,而是:面对具体问题时,应该优先考虑哪一类工具。这一点比单纯记软件名字更重要。
1)先看你研究的到底是哪一类体系
这是最基本的一步。如果你研究的是蛋白–小分子,那核心往往是口袋、构象搜索和打分排序;如果你研究的是蛋白–蛋白,那核心会转向界面互补、表面配对和聚类筛选;如果你研究的是蛋白–DNA/RNA,那又会进一步涉及骨架电性、碱基堆叠、二级结构以及核酸界面的专门特征。
也就是说,体系类型本身就决定了你不太可能只靠一个软件覆盖所有问题。
2)再看你是想“快速拿到初模”,还是想“做得更细”
有些工具更适合快速得到一个初始模型,有些工具则更适合做进一步 refinement。比如在蛋白–蛋白方向,HDOCK、ZDOCK、ClusPro往往更适合作为初筛或初始复合物生成工具;而 RosettaDock更适合放在候选界面已有雏形之后,继续做精修和优化。在蛋白–小分子方向,Vina往往是高效、常用的开源起点,而像 Glide、MOE、DS这类平台则更适合放到更完整、更工业化的工作流里理解。
3)还要看你手里有没有实验信息可以利用
如果你已经有一些实验线索,比如突变位点、交联信息、NMR 扰动、已知界面残基,那么某些工具的优势会一下子变得更明显。这时候,像 HADDOCK这样的信息驱动型平台就会更有价值,因为它不是单纯盲搜,而是能把实验约束真正纳入对接。相反,如果你几乎没有先验信息,只是希望先广泛探索可能的结合模式,那么更偏全局采样或自动化搜索的路线往往更适合作为起点。
4)也要看你更需要开源工具,还是商业一体化平台
这一点在实际项目里很重要。像 AutoDock4、Vina、ADFR、ADCP这一类开源路线,优势通常在于可获得性高、社区成熟、可重复性容易建立;而 Schrödinger/Glide、MOE、Discovery Studio这类商业平台的优势,则更多体现在模块完整、流程集中、前处理和后分析衔接顺畅。
所以很多时候,软件选择并不只是方法学问题,也和团队已有软件环境、预算条件以及工作流程习惯直接相关。
5)最后要看你希望得到什么样的结果
有些项目只需要一个可用的初始复合物模型,用于后续分析或实验设计;有些项目则希望得到一个更细化、更能支撑发表或深入讨论的结合模型;还有些项目希望把 docking 放到更长的流程里,继续接上 分子动力学、重打分、自由能分析等进一步计算。
如果目标不同,对工具的要求也会不一样。一个适合快速初筛的软件,不一定最适合做高精度精修;一个适合全局界面探索的软件,也不一定最适合处理核酸专门界面;而一个很强的复合物预测模型,也不一定能替代所有传统 docking 场景下的可控建模需求。
五、总结
不同软件在不同体系里各有优势。做蛋白–小分子对接时,AutoDock 系列、Glide、MOE、Discovery Studio 这类路线最常见;做蛋白–蛋白对接时,HADDOCK、HDOCK、ZDOCK、ClusPro、GRAMM、RosettaDock、AF3 更值得关注;做蛋白–DNA/RNA 对接时,则需要更多考虑 HDOCK、NPDock、3dRPC、P3DOCK 和 AF3 这类兼顾核酸界面的工具。
所以,分子对接软件并不是按“名气大小”来选,而是按“你面对的是哪一类复合体系、你更需要哪一种方法优势”来选。真正合适的软件,不一定是最有名的那个,而是最匹配你问题的那个。
(仅供大家参考,如有遗漏,欢迎交流指正。)
参考文献