 夜雨聆风
夜雨聆风





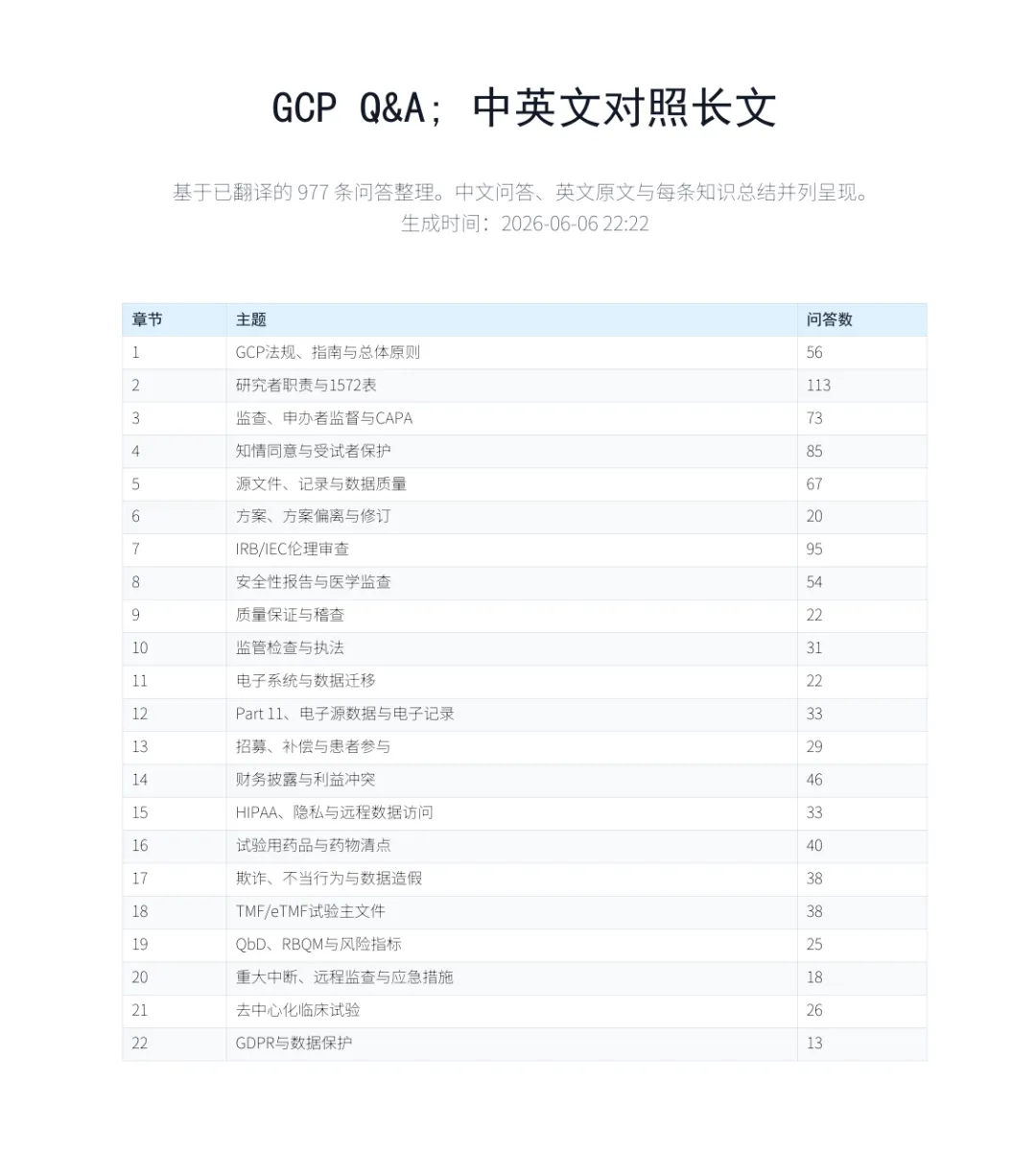
关于临床试验 977条常见问答, 附下载
本次整理的资料来源于 《Good Clinical Practice: A Question & Answer Reference Guide 2024-2025》。这是一部以问答形式编写的GCP实务参考书,内容覆盖FDA、ICH GCP、IRB/IEC、HIPAA、GDPR、RBQM、DCT、Part 11、安全性报告、财务披露、药物清点、TMF/eTMF等临床试验关键主题。相比单纯法规条文,这本书更接近真实项目中的问题场景:研究者能不能这样做?申办方该不该报告?IRB是否需要批准?监查员检查什么?哪些文件会在检查中被质疑?
为了方便国内临床研究、医学、运营、监查、质控、法规和伦理相关同事学习使用,我对全书问答进行了系统翻译和整理,并制作成中英文对照版本。每条问答均保留英文原文,同时配有中文翻译和简短知识总结,便于快速检索、学习和内部培训。部分内容请注意区分国内外监管差异。下载方式见文末
示例如下
6.10 Q. 纳入不符合入排标准的受试者,即使FDA和申办者批准,是否仍需IRB批准?
A. 通常需要IRB参与,很多情况下也需要IRB批准。研究者若希望纳入不符合入排标准的受试者,应先获得申办者批准,并提供充分记录的医学或科学理由,确保受试者安全不受损害。
但FDA和申办者批准并不足以替代IRB的伦理监督。IRB的职责是保护受试者权利和安全,应评估偏离请求的伦理影响、受试者安全风险和试验科学完整性。具体要求可能因机构政策和偏离性质而不同。
8.19 Q. 是否应把每个不良事件都先报告为可能与药物相关,直到证明不是?
A. 不应这样处理。研究者应根据现有证据和最佳医学判断,对SAE进行因果关系评估。21 CFR 312.32要求研究者立即向申办者报告任何SAE,并评估是否存在药物导致事件的“合理可能性”。
CIOMS建议对SAE采用二元判断:是否存在合理可能性认为IMP导致或促成该事件。非严重AE通常不要求研究者常规评估因果关系,除非方案规定为特别关注事件。评估应考虑时间关系、去激发/再激发、药理学一致性、同类药物经验、其他证据及替代解释。申办者最终仍需基于累计数据判断安全信号。
8.41 Q. 什么情况下可以对临床研究受试者揭盲?
A. 除非有令人信服的医学或安全理由,否则试验进行中不应揭盲。例如,了解治疗分配对处理严重不良事件必要时,可考虑揭盲。揭盲条件和程序应在方案中明确描述。研究者在揭盲前应尽量联系申办者医学监查员,共同讨论并决定。
申办者仅在有证据表明药物与不良事件存在合理可能因果关系时,才应对个别严重、意外、可疑不良反应进行揭盲。揭盲范围应尽量限制在临床研究团队之外的少数人。若揭盲后受试者为安慰剂组,通常不作为可疑不良反应报告;若为试验药物或阳性对照药,则需按IND安全报告或EudraVigilance要求报告。
10.15 Q:鉴于临床监查可以被视为一种内部审计活动,旨在确定临床研究者及其工作人员、伦理委员会以及其他各方是否正在履行其监管职责,那么根据机构政策,监查报告是否同样获得免于常规FDA检查的保护?
A. 不,主要区别在于临床监查是特定的FDA要求及申办方责任(21 CFR 312.56)。然而,这一现实并未阻止一些公司在FDA检查时主张监查报告应获得此类保护。
在2004年6月的警告信中,FDA引用了Biotronik, Inc.的违规行为,其中包括未“允许FDA调查人员检查和复制与检查相关的所有记录”,包括监查报告。警告信中写道:“你们拒绝在检查期间允许FDA调查人员直接查阅[研究]的监查报告,只允许检查与监查访视相关的信函文件”。“质量审计”或“内部审计”通常免于FDA审查,但FDA认为临床试验的监查报告不属于内部审计,因此在FDA检查期间不免除审查。申办者的责任包括确保对研究的适当监查,以确保遵守研究计划。由于未允许FDA检查和复制与检查相关的所有记录(包括监查报告),FDA调查人员无法确定该研究的监查是否足以确保研究者遵守研究计划。
当前的监查法规首次提出时,至少有一家公司认为,作为质量保证职能,如果不受政府审计,监查将更有效地运作。此外,该公司认为监查员的报告会更加坦诚和批判性,因此对申办者在确保根据需要采取适当的纠正措施方面更有价值。FDA拒绝了这一论点,表示FDA应保留根据第312部分检查与申办者临床研究监查相关记录和报告的权力。查阅这些材料有助于确认监查是否正在进行,并确定此类监查的性质。FDA也不认为检查监查记录和报告会显著影响监查员记录其观察和建议。
6.1 Q. 目前,在FDA生物研究监测检查中,方案相关问题被指为缺陷的情况有多常见?
A. 根据ICH E6(R3)草案,严格遵守临床方案对于确保临床试验数据的完整性和有效性至关重要。这与FDA检查发现一致:未遵守临床方案(未遵循研究计划)是CDER每年对临床研究者进行检查时最常引用的缺陷之一。
2023年临床试验场所FDA检查失败摘要显示,2018-2023年数据中有51%引用了未遵循研究计划。这些失败包括缺失知情同意文件、未执行方案要求的程序,和/或缺失IRB对研究变更审查的记录。该趋势已持续多年,反映出更广泛遵守ICH E6(R3)草案中GCP要求的必要性。
该指南强调方案遵守对数据完整性的重要性,这不仅包括方案偏离,也包括遵守FDA表格1572要求、病例研究记录准确性、以及对研究产品的充分追溯。此外,ICH E6(R3)草案强调受试者保护和知情同意的重要性,这些也是FDA检查中常见缺陷领域。这些方面的失败可能显著影响患者安全和试验结果可靠性。总体而言,FY2019年发布的国内检查报告中有超过50%引用了缺陷,提示需要加强GCP遵守,以提高临床试验质量和伦理标准。
后台回复GCPQA1 获取资料,包括QA英文原文-AI翻译-知识总结。