生成式 AI 可快速优化原料药的合成路线与晶型制备工艺,大幅提升规模化生产的收率、降低生产成本;同时可通过真实世界数据的学习,优化临床方案设计,精准定位受试人群,提升临床成功率。
据 Insilico Medicine 测算,生成式 AI 可将新药从靶点发现到 IND 申报的成本降低约 70%,这一成本优势,将彻底改变当前医药行业 “重磅药垄断” 的格局,为罕见病、未被满足临床需求的小众适应症药物研发打开全新空间。
2. 突破研发边界,打开难成药靶点的研发蓝海
当前全球获批的小分子药物,绝大多数靶向 GPCR、激酶等 “可成药靶点”,而占人类蛋白组绝大多数的 PPI 靶点、转录因子等,因结合界面平坦、无明确疏水口袋,传统小分子筛选极难获得高亲和力苗头化合物,被称为 “不可成药靶点”。而生成式 AI 的出现,彻底打破了这一壁垒。多肽分子可通过柔性结构与平坦的 PPI 界面实现高亲和力结合,是这类靶点的天然 “钥匙”;而生成式 AI 可精准提取多肽与靶点结合的关键药效团,将柔性的多肽结构转化为刚性、稳定、成药性更优的小分子骨架,让原本 “不可成药” 的靶点迎来了研发可能。这一技术路径,将未来小分子药物的研发边界,从现有不足 500 个可成药靶点,拓展到数千个潜在靶点,为肿瘤、自身免疫性疾病、神经退行性疾病等重大疾病的药物研发,提供了全新的源头创新方向。
3. 产业生态快速成熟,从单点工具到闭环研发体系
当前生成式 AI 在药物研发领域的应用,已从早期的 “单点工具试用”,进入到 “全流程体系化落地” 的阶段。一方面,以 AlphaFold、RFdiffusion 为代表的开源工具持续迭代,降低了行业应用门槛,全球高校、中小药企均可基于开源模型开展定制化研发;另一方面,跨国药企与 AI 制药企业的深度合作已成行业常态,礼来、诺华、默沙东等头部药企,均与晶泰科技、Insilico Medicine、Recursion 等 AI 企业达成数十亿美元级的合作,聚焦生成式 AI 在管线开发中的落地应用。据行业报告预测,2030 年全球 AI 制药市场规模将突破 1000 亿美元,其中生成式 AI 将占据核心份额。而随着干湿闭环的自动化实验室、机器人合成平台的普及,未来将形成 “AI 设计 – 自动化合成 – 高通量验证 – AI 迭代优化” 的全自动化研发管线,真正实现药物研发的智能化、工业化变革。
传统药物设计中,一个高频出现的问题是:计算机设计的高活性分子,在实验室中合成难度极高、收率极低,甚至无法合成,导致研发投入付诸东流。这也是早期计算机辅助药物设计(CADD)始终无法完全落地的核心痛点之一。而新一代生成式 AI 模型,已将合成可及性作为核心约束条件纳入分子生成环节。例如 DeepCubist 模型在生成拟肽小分子时,会优先选择合成路线成熟、原料易得的分子骨架与官能团,确保生成的分子具备工业化合成的潜力;行业内的 CACHE 挑战等基准测试,也已将 “按需合成” 作为核心评价指标,推动生成式模型从 “设计可结合的分子” 向 “设计可合成、可成药的分子” 升级。合成可及性的前置把控,大幅减少了后期合成失败的风险,降低了原料药的生产成本,让 AI 设计的候选分子,真正具备从实验室走向工业化生产的落地能力。
5. 解锁难成药靶点的研发潜力,填补行业空白
PPI 等难成药靶点,是当前全球新药研发的蓝海,也是未被满足临床需求最集中的领域。传统小分子高通量筛选,在这类靶点上的苗头化合物检出率不足 0.1%,而多肽筛选可轻松获得纳摩尔级亲和力的结合剂,是这类靶点最有效的研发起点。生成式 AI 的出现,彻底打通了 “多肽高亲和力结合剂→小分子成药候选物” 的转化通道,让原本无法通过传统方法获得小分子苗头的靶点,迎来了研发突破的可能。例如,针对多个肿瘤相关的 PPI 靶点,已有研究团队通过 AI 生成的多肽结合剂,成功转化得到了高活性的小分子抑制剂,填补了靶点研发的空白。这一技术路径,将彻底改变当前小分子药物研发靶点高度同质化的行业现状,推动源头创新药物的爆发。
三、生成式 AI 在药物研发领域的标杆落地案例
从基础研究到产业落地,生成式 AI 在药物研发领域已诞生了大量可验证、可复制的成功案例,其中既有 “多肽苗头→小分子先导” 的专项突破,也有全流程药物研发的里程碑式成果,充分验证了这一技术的产业化价值。
(一)多肽苗头到小分子先导转化的核心研究与应用案例
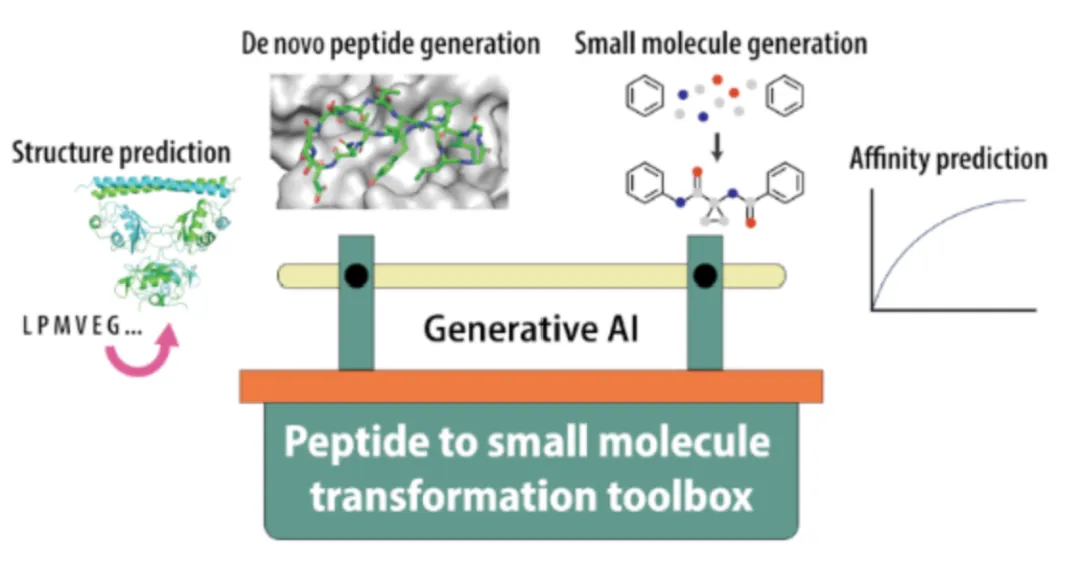
1. 悉尼大学全流程 AI 管线:实现多肽到小分子的端到端转化
2026 年,悉尼大学 Joshua Mills 与 Yu Heng Lau 团队在《Beilstein Journal of Organic Chemistry》发表的研究,是当前生成式 AI 在多肽 – 小分子转化领域最系统的成果。团队构建了覆盖全流程的 AI 工具集,实现了从靶点到小分子先导的端到端赋能:
英矽智能开发的 INS018_055,是全球首个由生成式 AI 全程设计、进入临床阶段的小分子药物,用于治疗特发性肺纤维化(IPF)。该药物从靶点发现到先导化合物优化,全程由英矽智能的生成式 AI 平台完成,研发周期从传统的数年压缩至 18 个月,研发成本降低超 70%。目前 INS018_055 已完成 II 期临床,展现出优异的安全性与有效性,成为生成式 AI 在新药研发领域的里程碑事件,验证了生成式 AI 全流程药物研发的可行性。
华盛顿大学 David Baker 团队开发的 RFdiffusion 扩散模型,是生物大分子生成领域的标杆性成果。该模型可基于靶点结构,从头设计高亲和力的蛋白、多肽结合剂,设计效率与成功率远超传统理性设计方法。基于 RFdiffusion,团队已成功设计出新型抗肿瘤蛋白药物、多肽疫苗、PPI 靶点的多肽抑制剂,相关成果连续发表于《Nature》《Science》等顶刊。目前该模型已开源,成为全球多肽药物研发的核心工具,为 “多肽优先” 的药物设计策略提供了源头支撑。
4. Recursion Pharmaceuticals:AI 驱动罕见病药物研发
Recursion Pharmaceuticals 是 AI 制药领域的头部企业,其基于生成式 AI 与高通量生物实验平台,构建了全球最大的细胞表型数据库,专注于罕见病药物的研发。通过生成式 AI 对疾病表型与分子活性的学习,Recursion 已发现了多个针对罕见病的全新候选药物,其中针对脑小血管病的 REC-994、针对神经纤维瘤病的 REC-4881 等多个分子已进入临床阶段,验证了生成式 AI 在罕见病药物研发中的独特优势 —— 解决了罕见病靶点研究少、传统筛选成本高的行业痛点。
四、挑战与未来展望
尽管生成式 AI 在药物研发领域已展现出颠覆性的价值,但行业仍面临着亟待突破的核心挑战。其一,训练数据的质量与覆盖度仍有不足,小分子化学空间的多样性极高,而现有高质量的蛋白 – 配体结合实验数据仍相对稀疏,尤其是小众靶点、多肽 – 靶点复合物的数据不足,限制了模型在部分场景的泛化能力;其二,AI 模型的 “可解释性” 仍需提升,当前多数生成模型属于 “黑箱模型”,无法完全解释分子生成与活性预测的内在逻辑,一定程度上影响了药化专家的接受度与研发决策;其三,干湿闭环的验证体系仍需完善,AI 模型的预测结果,最终仍需通过实验验证,而当前 AI 设计与实验验证的联动仍未完全实现自动化,制约了研发效率的进一步提升。但不可否认的是,生成式 AI 已经不可逆地改变了药物研发的底层范式。未来,随着模型的持续迭代、高质量实验数据的积累、自动化干湿闭环平台的普及,生成式 AI 将实现三大核心突破:第一,端到端全自动化研发管线的落地,实现从靶点验证到 IND 申报的全流程 AI 驱动,将新药研发周期从 10 年压缩至 3-5 年,让更多创新药快速进入临床;第二,多肽优先策略成为难成药靶点研发的主流范式,随着多肽 – 小分子转化 AI 工具的持续成熟,PPI 等难成药靶点将迎来研发爆发,填补大量未被满足的临床需求;第三,药物研发的民主化与普惠化,开源 AI 工具的普及,将打破头部药企对新药研发的垄断,让高校、中小药企、科研机构均可开展源头创新,推动全球医药产业的多元化发展。生成式 AI 从来不是要替代药化专家、结构生物学家与临床研发人员,而是成为研发人员的 “超级助手”,将科研人员从重复、繁琐的试错工作中解放出来,聚焦于更具创造性的科学问题。在生成式 AI 的赋能下,新药研发将从 “劳动密集型的试错产业”,转变为 “数据驱动的智能创造产业”,而 “从多肽苗头到小分子先导” 的技术突破,正是这场产业变革中最具代表性的缩影。[1] Mills J, Lau Y H. Using generative AI to transform peptide hits into small molecule leads[J]. Beilstein Journal of Organic Chemistry, 2026, 22: 672-679. https://doi.org/10.3762/bjoc.22.51[2] Chen C, Zhang X, Du H, et al. Enantioselective Skeletal Transformation of 1,2,3-Thiadiazoles to Dihydrothiophenes[J]. The Journal of Organic Chemistry, 2026, 91, 551-556. https://doi.org/10.1021/acs.joc.5c02611[3] Dong B, Li X, Gao G, et al. Practical and Scalable Manufacturing Process for Sparsentan. Part 1: Development of a Robust and Economical Route to a New Benzyl Alcohol Intermediate[J]. Organic Process Research & Development, 2026. https://doi.org/10.1021/acs.oprd.5c00412[4] Igashov I, Stärk H, Vignac C, et al. DiffLinker: Equivariant 3D conditional diffusion model for molecular linker design[J]. Nature Machine Intelligence, 2024, 6(4): 417-427. https://doi.org/10.1038/s42256-024-00815-9[5] He X, Zhang Y, Lin H, et al. Peptide2Mol: Pocket-aware diffusion model for transforming peptide binders to small molecules[J]. arXiv preprint arXiv:2511.04984, 2025. https://doi.org/10.48550/arxiv.2511.04984[6] Adams K, Abeywardane K, Fromer J, et al. ShEPhERD: Interaction-aware diffusion model for bioisosteric fragment merging[J]. arXiv preprint arXiv:2411.04130, 2024. https://doi.org/10.48550/arxiv.2411.04130[7] Umedera K, Yoshimori A, Chen H, et al. DeepCubist: a deep learning-driven molecular generator for peptidomimetic design[J]. Journal of Computer-Aided Molecular Design, 2023, 37(2): 107-115. https://doi.org/10.1007/s10822-022-00493-y[8] Passaro S, Corso G, Wohlwend J, et al. Boltz-2: Joint structure and affinity prediction for protein-ligand complexes[J]. bioRxiv preprint, 2025. https://doi.org/10.1101/2025.06.14.659707[9] Zhou G, Rusnac D V, Park H, et al. RosettaVS: AI-accelerated virtual screening with physics-based docking[J]. Nature Communications, 2024, 15(1): 7761. https://doi.org/10.1038/s41467-024-52061-7[10] Ackloo S, Al-awar R, Amaro R E, et al. The Critical Assessment of Computational Hit-finding Experiments (CACHE) initiative to benchmark computational methods for hit-finding[J]. Nature Reviews Chemistry, 2022, 6(4): 287-295. https://doi.org/10.1038/s41570-022-00363-z[11] Pelay-Gimeno M, Glas A, Koch O, et al. Structure-based design of non-peptidic peptidomimetics[J]. Angewandte Chemie International Edition, 2015, 54(31): 8896-8927. https://doi.org/10.1002/anie.201412070
 夜雨聆风
夜雨聆风