 夜雨聆风
夜雨聆风





一篇搞懂:OpenClaw如何让计算化学“自己跑起来”?

模型工具概述:OpenClaw + 技能库 = 会自己干活的代理
传统的计算化学自动化要么靠死板的工作流(比如AiiDA、FireWorks),要么靠专门训练的大模型代理(比如ChemCrow)。但这篇文章提出了一个更灵活的设计:通用代理 OpenClaw 做总控,领域技能(Skills)干具体活。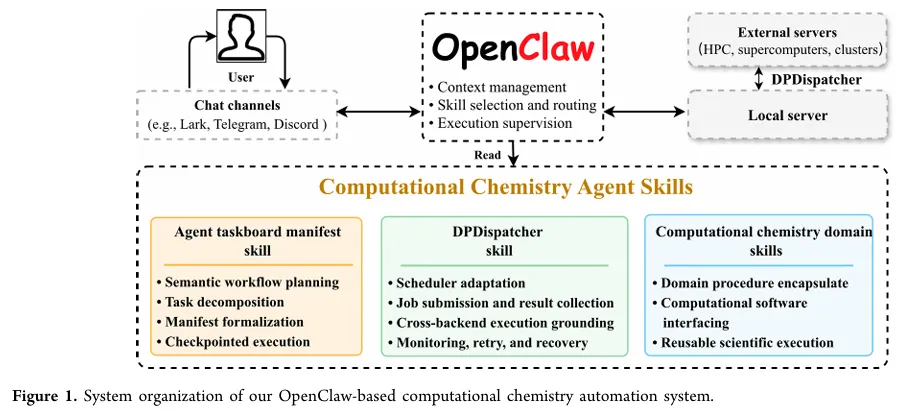
核心模块包括:
- Agent Taskboard Manifest Skill
:把自然语言任务翻译成结构化的工作流清单(阶段、依赖、输入输出、验证条件)。懒加载策略让代理只关注当前子任务。 - DPDispatcher Skill
:基于开源DPDispatcher,统一对接Slurm、PBS、LSF等调度器,自动生成脚本、提交、监控、取结果。 - 计算化学技能库
(LGPL-3.0开源):覆盖量子化学(Gaussian, VASP, CP2K)、分子动力学(LAMMPS, Amber)、机器学习势(DeepMD-kit)、分析工具(Phonopy, ReacNetGenerator)等。每个技能就是一个可执行命令+依赖描述,通过 uvx隔离运行。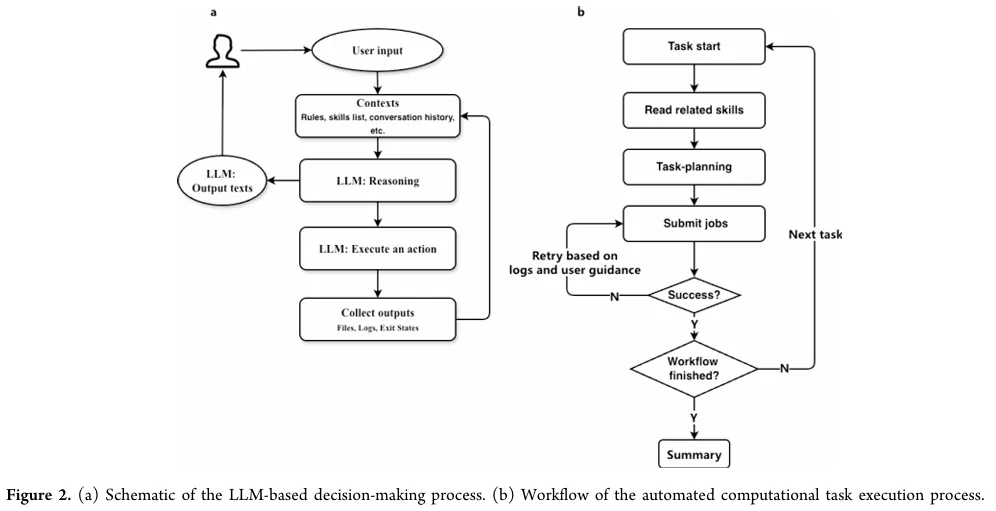
这种设计的好处是:新增软件只需写一个新技能,不用改代理内核。而且代理的上下文不会爆炸——需要用哪个技能才加载哪个。
用途实证:甲烷氧化反应MD的全程自动跑通
为了验证这套系统能不能真的干活,作者复现了之前一篇 Nature Communications 上的甲烷氧化MD研究。输入一句自然语言指令(包含优化、建盒、跑DP势、升温、1 ns反应MD、ReacNetGenerator分析),系统自动拆解为6个阶段:
-
分子结构准备(Open Babel) -
几何优化(Gaussian, B3LYP/6-31G(d,p)) -
格式转换(dpdata) -
反应体系构建(Packmol:50 CH₄ + 100 O₂,密度0.25 g/cm³) -
反应MD(LAMMPS + DeePMD-kit,3000 K NVT,1 ns,步长0.1 fs) -
轨迹分析(ReacNetGenerator提取反应路径) 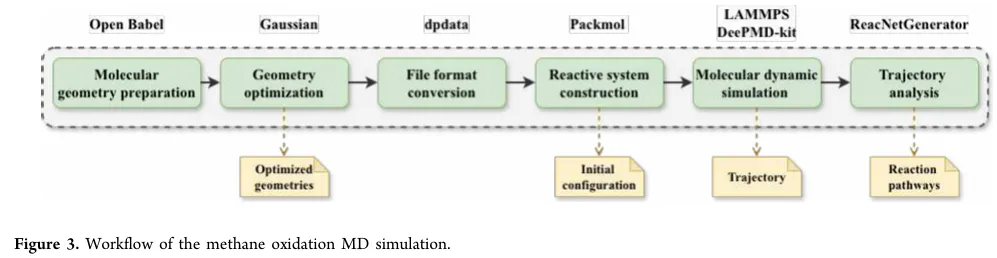
关键亮点:
- 预检
:执行前检查可执行文件、依赖、调度器权限,避免环境问题。 - 状态驱动
:只有前一步输出通过验证,才进入下一步。 - 错误恢复
:代理读取日志、返回码,自动尝试参数修复、重试、回滚。实在不行才请求人工介入。 - 可复现性
:所有输入文件、参数、中间结果都有记录(见补充信息)。
最终成功跑出CH₄和O₂的消耗曲线,以及H₂O、CO、CO₂的生成趋势(图4)。即使LLM存在随机性(文中用GPT‑5.5),系统仍能通过反馈闭环完成任务。
📊 一次典型运行消耗:462,662输入token + 76,129输出token + 6M缓存token(GPT‑5.5 API)。
小编总结
-
OpenClaw+技能库的设计,让计算化学自动化从“写死的工作流”升级为“可推理、可纠错、可插拔”的智能代理。 -
你不需要重写代理,只要新增或替换一个技能(比如换个量子化学软件),就能扩展能力。 -
虽然代理能跑通流程,最后的科学有效性判断仍需人类专家(检查模型、参数、采样是否合理),但重复的“脏活累活”可以彻底交给它了。 点击下方链接-下载原文pdf

automating-computational-chemistry-workflows-via-openclaw-and-domain-specific-skills.pdf
往期好文推荐

往期课程推荐

