 夜雨聆风
夜雨聆风





SRR数据的下载与多线高效程处理
一.什么是SRR数据
-
研究层次(Study):常以SRP为起始标志,代表一个研究项目。 -
样本层次(Sample):常以SRS为起始标志,代表产生数据的生物样本。 -
实验层次(Experiment):常以SRX为起始标志,用于记录相关实验设计。 -
运行层次(Run):常以SRR为起始标志,代表测序数据,是我们直接下载和处理的数据。各 SRR数据均有多个“spot”,各spot代表一次测序产生的数据。在双端测序中,各 spot 由两条读段(read,及常见的R1和R2),本文后续介绍的fasterq-dump程序在运行的过程中需要加入–split-3 参数,其目的即是为了处理R1和R2的正确分配。
二.如何获取SRA数据相关信息
2.1.网页一:SRA Explorer
2.2.网页二:SRA Run/File Selector (个人觉得更好用)
三.需要用到的软件
3.1.sratoolkit
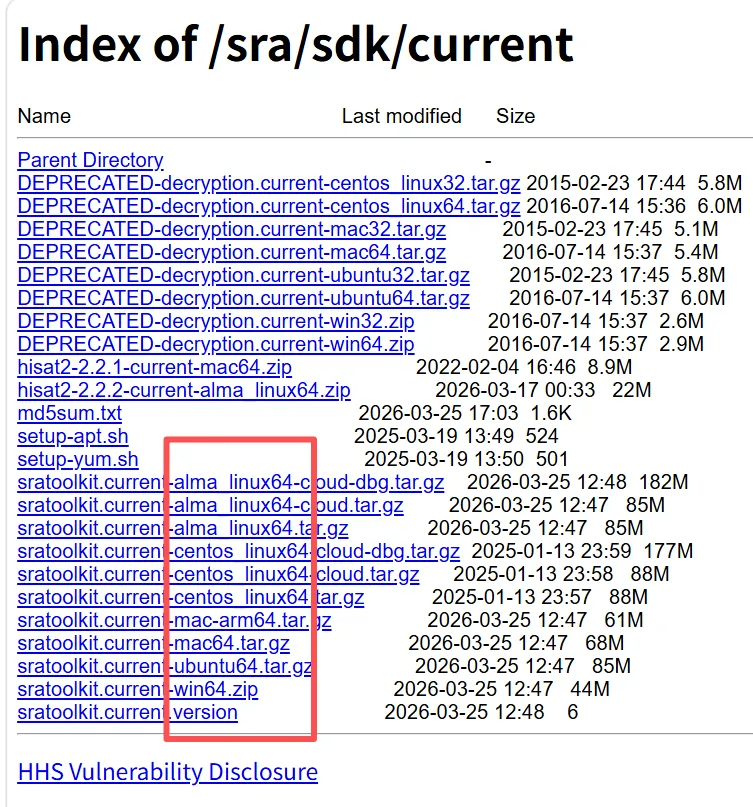
3.1.1.conda安装
3.1.2.手动安装(建议手动安装)
mkdir ~/software #在家目录下建立软件文件夹,如果已有,可忽略cd software #进入软件文件夹#下载sratoolkitwget -c -b https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/3.4.1/sratoolkit.3.4.1-alma_linux64.tar.gz#解压即用tar -zxvf sratoolkit.3.4.1-alma_linux64.tar.gz#将软件加入环境变量,注意用追加(>>),而不是覆盖(>)重定向。如果不将软件加入环境变量,需用绝对路径调用软件echo 'PATH=$HOME/software/sratoolkit.3.4.1-alma_linux64/bin:$PATH' >>~/.bash_profile. ~/.bash_profile #重新激活环境,即可用相关软件
3.2.pigz
四.数据下载与处理
4.1.基于SSR号的prefetch法
4.1.1.输入项目号并点击search。
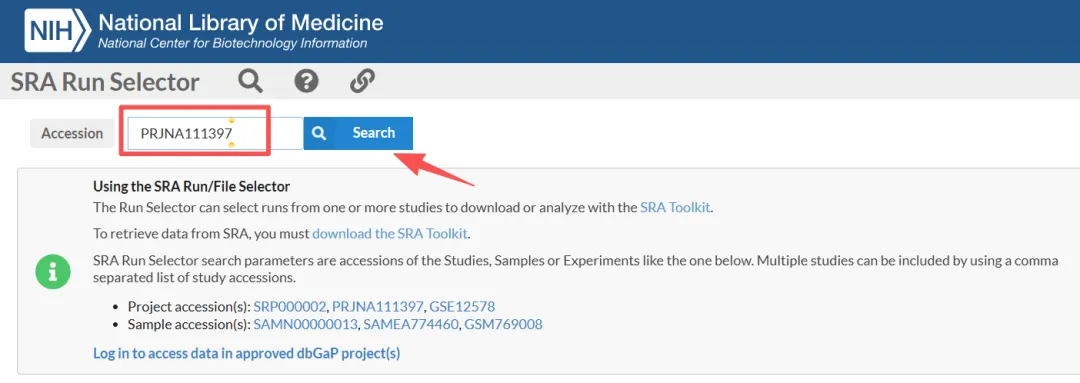
4.1.2.点击Metadata或Accession获取SRR序列号
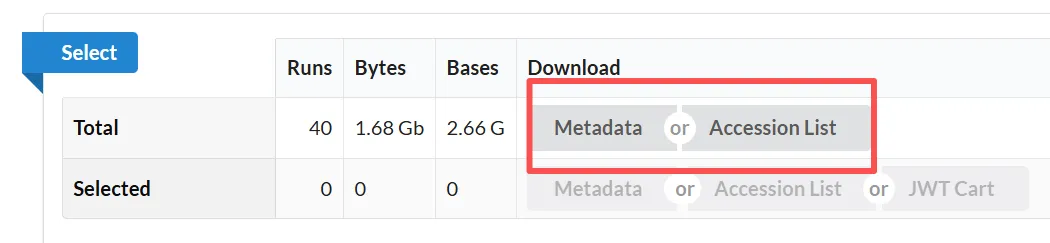
4.1.3.批量下载数据(取2个srr数据做演示)
mkdir PRJNA111397 && cd PRJNA111397 #建立并进入数据文件夹for srr in SRR013550 SRR013551do #-X限定可下载的SRR文件的大小,默认大小只有20G,如果服务器线程不够,不要一次性把SRR号输入到for行后面,可分批次输入和下载prefetch -X 500GB $srr >log.$srr.prefetch 2>&1 &done#下载完成后须检测log文件,查看下载是否成功(具HTTPS download succeed字样),如果失败(具failed或error字样,也可能是别的标识),需重新下载。
4.1.4.将srr数据批量转换为fastq(多线程高效)
for srr in SRR013550 SRR013551dofasterq-dump $srr --split-3 >log.$srr.fasterqdumpdone
4.1.5.批量压缩
for srr in SRR013550 SRR013551dopigz -p 10 $srr*.fastq #-p指定线程数done
4.1.6.确认文件下载无误后可删除SRR文件以节约空间
rm -r SRR*4.2.基于文件地址(prefetch下载失败时可用该方法)
4.2.1.在BioProject数据库查找相关信息
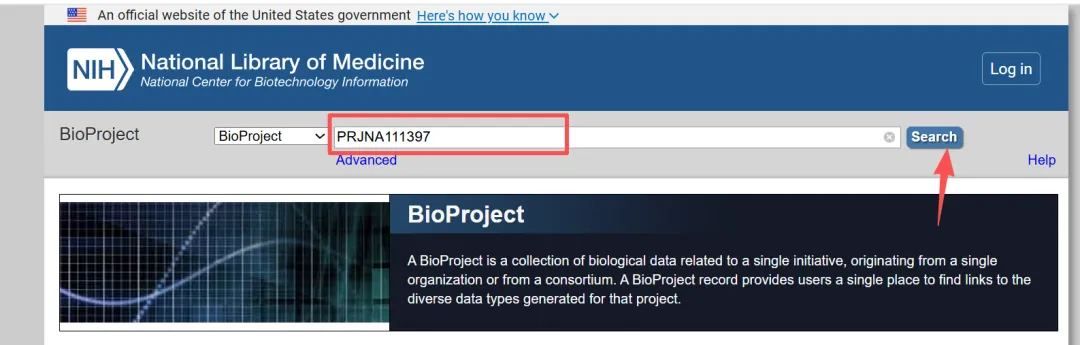
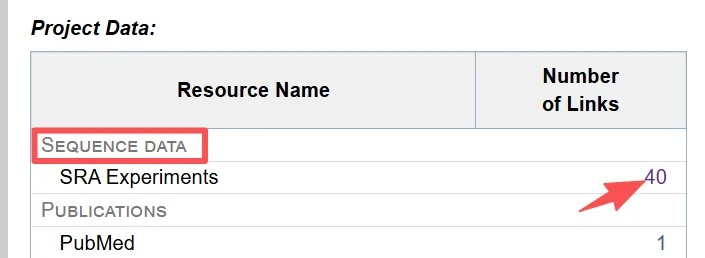
4.2.2.点击测序数据SRA Experiments数目进入数据详情页
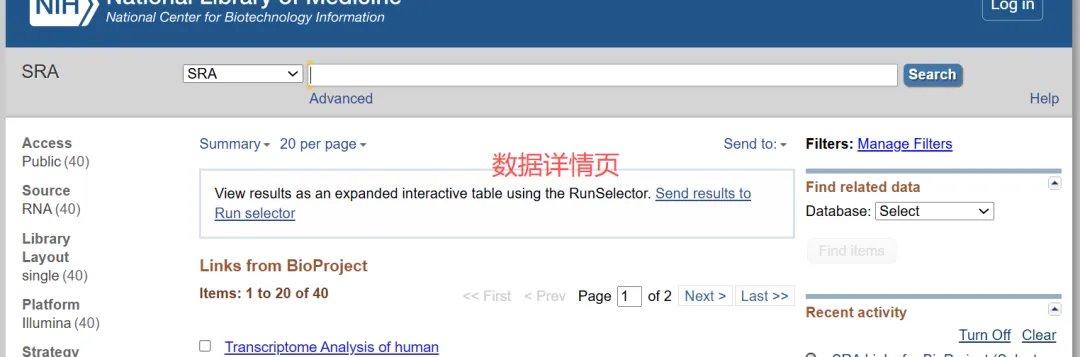
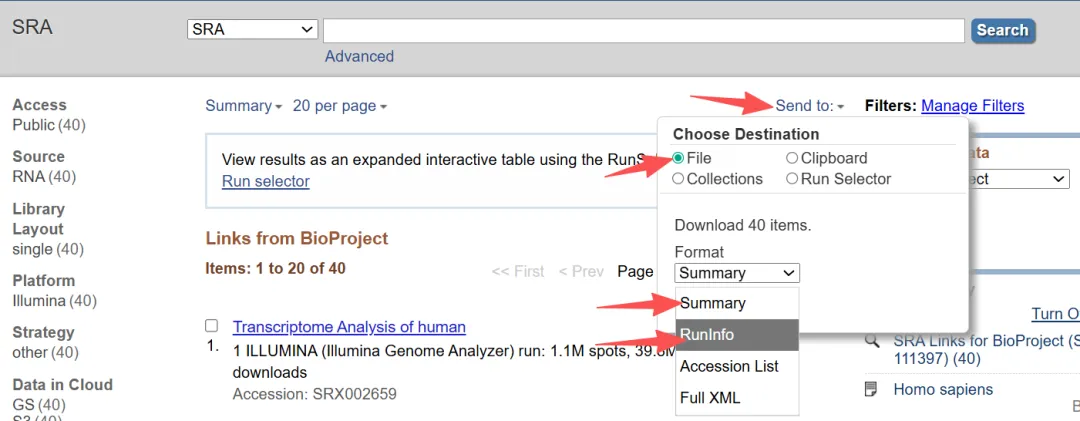
4.2.3获取srr文件地址
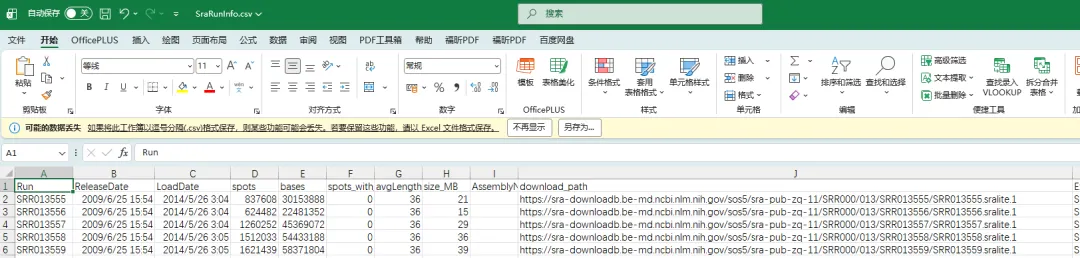
4.2.4.利用wget下载数据和后续处理
mkdir PRJNA111397 && cd PRJNA111397 #建立数据文件夹并进入#数据下载wget -c -b https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos5/sra-pub-zq-11/SRR000/013/SRR013550/SRR013550.sralite.1wget -c -b https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos5/sra-pub-zq-11/SRR000/013/SRR013551/SRR013551.sralite.1#for循环处理数据,这个是直接用文件名for srr in SRR013550.sralite.1 SRR013550.sralite.1dofasterq-dump -e 10 $srr --split-3 >log.fstrq.dump.$srr 2>&1 & #srr转fastqdonepigz -p 50 SRR*.fastq >log.pigz.fastq.pigz 2>&1 & #压缩文件mkdir srrmv SRR* srr/mv srr/*.fastq .rm srr -rf #确认数据无误后删除srr文件