 夜雨聆风
夜雨聆风





分子对接)从软件安装到结果分析与制图
点击蓝字 关注我们

今天分享的是
分子对接完整教程
分子对接简介
Introduction to Molecular Docking
分子对接(Molecular Docking):是通过分子的特征以及分子和分子之间的相互作用方式来进行计算的一种理论模拟方法,在材料、化学、生物和物理等专业领域均有应用,蛋白质、小分子、晶体材料等都可以进行分子对接。
分子对接技术在生物医药领域的应用最为广泛,可以将其解释为一种通过受体的特征以及受体和药物分子之间的相互作用方式来进行药物设计的理论模拟方法,主要用来研究分子间(如配体和受体间)的相互作用,并预测其原子水平上的结合模式和亲合力,从而评估药物的活性、选择性等,解析药物与蛋白之间相互作用的基本生化过程。
分子对接的原理与方法
分子对接的原理
微观尺度的原子或分子同样遵守牛顿定律、法拉第定律及热力学定律等物理、化学定律,通过这些物理、化学定律可以对原子或分子进行建模计算,如对小分子药物与靶标蛋白的结合过程进行模拟和计算,获取这一过程的自由能变化,从而预测小分子药物与靶标蛋白的结合。
空间匹配是分子间发生相互作用的基础,能量匹配是分子间保持稳定结合的基础。具体来说锁钥模型和诱导嵌合学说是分子对接的理论基础,分子间相互作用力如氢键、盐键、范德华力、静电作用和疏水作用等为分子对接提供了能量驱动。
分子对接的方法
柔性对接方法:对接过程中允许配体和受体的构象发生自由变化,但由于变量随着整个体系的原子数呈现几何级数增长,因此柔性对接方法的计算量非常大,消耗计算机时很多,具体操作过程中一般只指定蛋白结合口袋里的少数关键残基构象可变,适用于精确考察分子间识别情况。如果要研究更大程度的构象变化,需要用到分子动力学(Molecular Dynamics ,MD)模拟进行分析。
刚性对接方法:所谓刚性对接,即参与对接的配体和受体分子构象不发生变化,仅改变空间位置与姿态。刚性对接方法的简化程度最高,计算量相对较小,适合于进行大分子之间的对接、高通量虚拟筛选研究。
半柔性对接方法:半柔性对接方法允许对接过程中小分子(配体)构象发生一定程度的变化,但通常会固定大分子(受体)的构象,即将受体分子视为刚性的,另外小分子构象的调整也可能受到一定程度的限制,如固定某些非关键部位的键长、键角等,而通过平动、转动及可旋转二面角扭转等产生多种构象。半柔性对接方法兼顾计算量与模型的预测能力,是应用较多的对接方法之一。
分子对接的具体应用
生物医药领域
分子对接技术在生物医药领域的应用最为成熟和广泛,主要包括药物分子的虚拟筛选、靶点的筛选验证、药物递送系统的开发、抗菌/抗病毒材料的功能验证等。
抗菌/抗病毒复合材料的作用机制研究:如Dana等采用绿色、经济的方法合成了一种具有抗菌效能的ZnO/儿茶素纳米复合材料,并利用分子对接技术将从绿茶中提取的天然染料儿茶素(B)以及三种构型的纳米复合材料(C1)、(C2)、(C3)分别与大肠杆菌24kDa结构域-抗生素氯新生霉素(Clorobiocin)复合物的晶体结构(PDB ID为1KZN)进行了对接,对接结果显示这些化合物能够紧密占据1KZN的结合腔,其中纳米复合材料C1和C2尤其具备在活性位点实现稳定结合的能力。
https://doi.org/10.1016/j.matchemphys.2024.129408
环境/材料领域
分子对接技术在环境和材料领域也有一定的应用,如手性MOFs材料对映体识别能力研究、生物质材料性能改进、金属有机框架(MOF)筛选等。
手性MOFs分离材料研究:宋睿科等合成了一种手性金属有机骨架(钴-甘氨酰-L-谷氨酸,Co-L-GG),并将Co-L-GG作为手性功能单体通过一步原位聚合法制备手性毛细管整体柱(手性金属有机骨架修饰毛细管硅胶整体柱CMOF-SMCC)。随后将Co-L-GG作为受体分子,手性药物作为配体分子进行了分子对接,获得了Co-L-GG与四种与分离度较好的手性药物的结合自由能、结合自由能差、对映体选择性因子、分离度和分子间相互作用(范德华力除外)等关键信息,使用Discovery studio分析了主体-客体复合物构型,揭示了CMOF-SMCC对手性药物的识别机理。
DOI: 10.3724/SP.J.1123.2024.01020
食品科学领域
分子对接技术在食品相关研究中的应用也较为丰富,主要集中在食品功能性成分挖掘与活性机制解析等方面。
食品中功能肽的筛选、开发:例如zhao等通过虚拟筛选、分子对接和电子舌分析等技术鉴定了来自虹鳟的鲜味肽,具体应用是选取了332种具良好水溶性和无毒特性的肽与鲜味受体T1R1/T1R3(异二聚体)进行分子对接,根据CDocker-Energy的高低(越低表明亲和力更强),筛选出了20种肽,又根据鲜味肽呈鲜的结构组成特点选取并合成了三种四肽即EANK、EEAK和EMQK。随后再次利用分子对接技术对这三种四肽和鲜味受体T1R1/T1R3的互作机制进行了研究,结果表明肽EANK、EEAK和EMQK可以进入T1R1腔的结合口袋,氢键和静电相互作用是重要的相互作用力,且发现氨基酸残基Arg277、Arg151、Asp147和Gln52可能在鲜味的产生中起关键作用。
https://doi.org/10.1016/j.fshw.2022.07.026
分子对接的详细步骤
Detailed Steps of Molecular Docking
基于自身专业背景,本期分子对接教程主要围绕蛋白质和小分子配体化合物的对接开展,因此部分表述有一定专业倾向性。不同专业领域分子对接方法和流程有一些细微的差异,同时因个人的喜好和习惯不同,具体使用的分子对接工具可能也会有所出入。
工作环境的准备
Python环境的准备
Python:AutoDock和PyMol都是基于Python编程语言的软件,因此在下载前需要准备好python编译环境。
(1)官方网址:https://www.python.org/
(2)Python的安装步骤:目前最新版本的分子对接软件AutoDock vina等不再需要单独下载Python配置编译环境。为了满足不同版本用户的需求,这里还是简单介绍一下python的安装步骤①访问官方网站,点击”downloads“;②点击电脑所用系统,选择需要的版本进行下载;③按照指引完成安装即可。
注:如果安装的是最新几个版本的分子对接软件,这一步可以跳过。
AutoDock Vina的准备
AutoDock Vina:由美国Scripps研究所Oleg Trott博士于1980年代末开发,是目前最快速且使用最广泛的开源免费分子对接软件之一,现如今主要由Scripps研究所的Forli实验室负责软件的维护和开发。
(1)官方网址:https://autodock.scripps.edu/
(2)AutoDock vina的安装步骤:①访问官方网站;②点击“PROJECTS”,选择“Docking”;③Computational Docking Software栏目下可以选择不同的软件版本,点击软件简介中的名称可以进入软件详细介绍界面;④点击左侧菜单栏“Downloads”进入下载界面;⑤选择与自己电脑系统匹配的版本并点击“Files”栏目下的软件名称进行下载;⑥下载好后直接点击运行下载好的驱动文件;⑦点击“next”;⑧点击“I Agree”再点击“next”;⑨选择一个保存地址,尽量存到D盘,给文件夹命名如“AUTODOCK”,点击“next”;⑩继续点击“next”进行安装,注意这时候安装好的是几个可执行文件,还不能正常运行使用;⑪接下来还需要添加环境变量,复制安装好的可执行文件所在文件夹的存储路径如“D:\AUTODOCK”,然后在“文件资源管理器”或桌面选则“此电脑”,单击右键选择“属性”→“高级系统设置”→“环境变量”→选中“Path”变量并点击“编辑”,然后再点击“新建”将前面复制的路径“D:\AUTODOCK”粘贴进来,连续点击三个“确定”即可;⑫检查软件是否安装成功,“Windows+R”,输入“cmd”,回车,输入vina,“回车”,正常运行则说明软件安装成功。
(3)常见安装问题及解决方案:在安装AutoDock Vina时普遍遇到的问题是打开Windows版本下载链接显示乱码,无法执行下载程序。利用deepseek进行分析,给出的其中一个解决方案为“尝试直接使用右键菜单将下载网址另存到桌面”,实践证明该方法有效,并根据结果追问倒查,得到的回答是因“服务器返回HTTP头不正确”造成的错误(仅供参考)。具体操作步骤:①将鼠标光标定位到需要下载的软件版本名称超链接,注意不要打开;②鼠标光标变为手形时,单击右键并选择“将链接另存为”;③选择存储地址,存到桌面即可。除此之外,也可选择“绕道而行”,即通过github平台获取安装驱动文件。
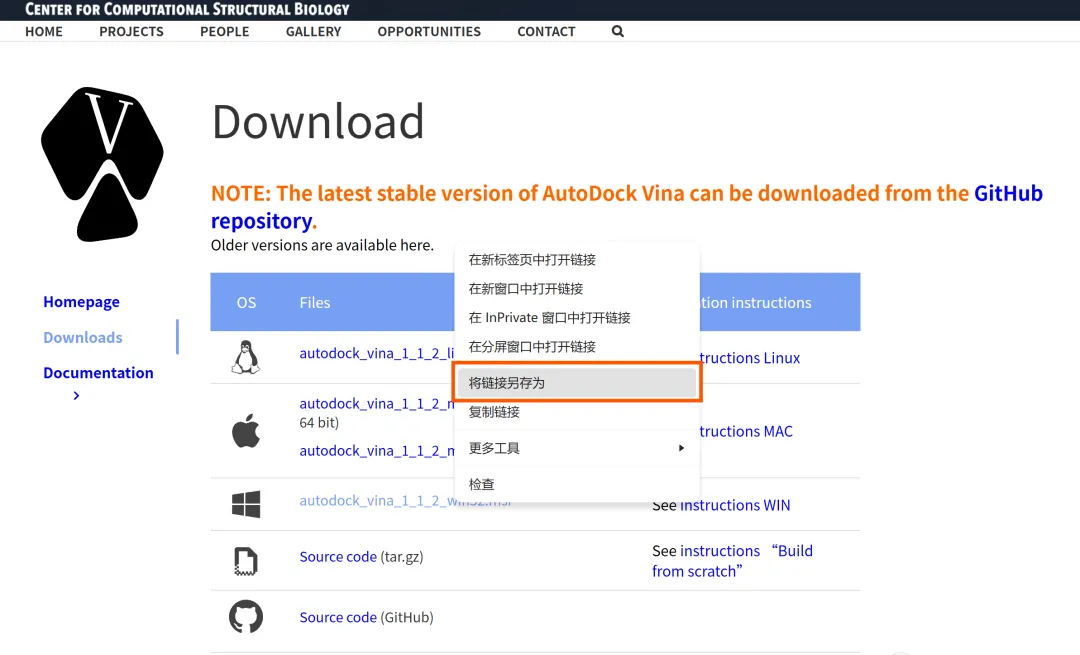
图 1 将Windows版本的软件下载链接(驱动)另存到桌面
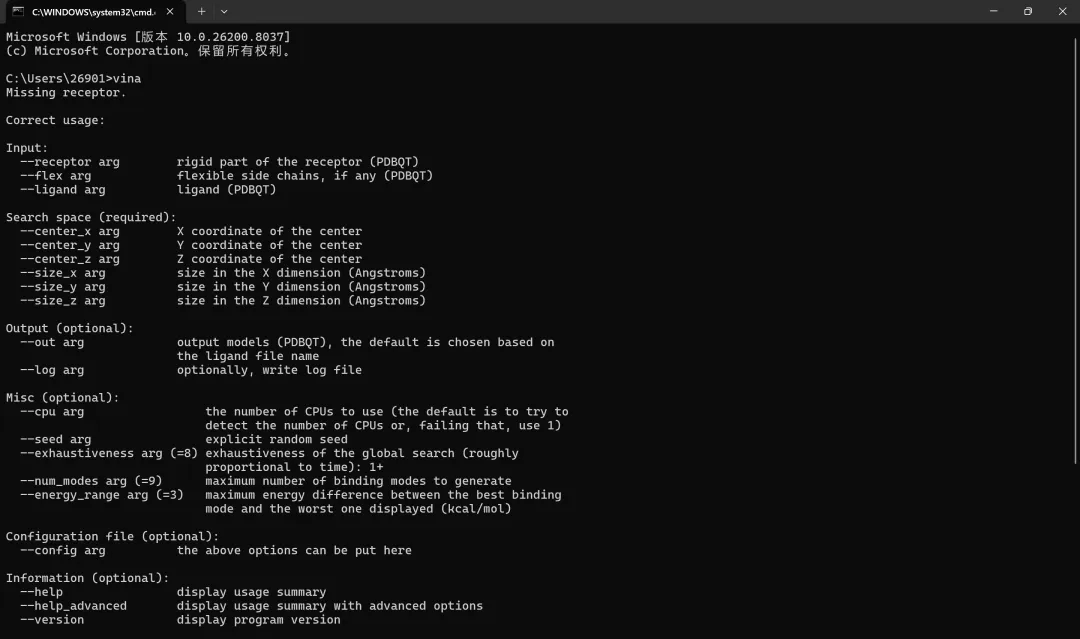
图 2 输入vina返回该结果表明AutoDock vina安装成功
注:AutoDock vina没有软件图标,也没有具体的可视化操作界面,只要输入命令后返回的结果没问题即可。
MGLTools的准备
MGLTools:由Scripps研究所开发的用于分子结构可视化与分析的软件套件,一般只用到其中的AutoDockTools来处理蛋白质和小分子文件,包括去水、去金属离子和加氢等处理,以支持AutoDock vina分子对接程序。
(1)官方网址:https://ccsb.scripps.edu/mgltools/
(2)MGLTools安装步骤:①访问官方网站,点击左侧菜单栏的“Downloads”,选择并点击需要的版本下载安装包(推荐下载1.5.6版本);②下载完成后点击安装程序进行安装,点击“next”;③勾选“I accept the…”,点击“Yes”;④点击“Browse”选择安装位置,最好安装在D盘,且最好保证安装路径不存在中文,命名文件夹如“MGL”,点击“next”进行安装,点击“ok”安装配套的几个组件,再根据需要勾选是否创建快捷方式等选项,然后点击“Finish”即可。⑤把安装好的文件夹中的名称为“adt”的“windows批处理文件”复制到D:\AUTODOCK路径的文件夹中,再双击该文件即可进入软件,也可通过双击桌面图标打开。
(3)常见安装问题及解决方案:在MGLTools安装中出现频次最高的问题是软件安装完成后,打开AutoDockTools的加载过程中出现软件闪退问题,通过查阅各方资料和询问deepseek均未得到具体的解决方案,但是获得了一个大致的问题方向“ADM Ryzen显示适配器AMD Radeon(TM) Graphics核显与软件不适配(该问题一般只出现在Ryzen用户中)”,因此解决思路是“禁用该显示适配器”,注意是禁用而非删除,网上有的教程会告诉你直接将其删除,虽然软件可以正常使用了,但是当你今后需要用到该适配器的时候,重装驱动是一件极其麻烦的事情,所以不建议直接将适配器删除。具体禁用步骤:①在文件资源管理器或桌面选中“此电脑”,单击右键选择“显示更多选项”,然后再点击“管理”进入计算机管理界面;②点击“系统工具”下的“设备管理器”;③下翻找到“显示适配器”,打开下拉选项选中“AMD Radeon(TM) Graphics”;④单击右键选择“禁用设备”,这时候再打开MGLTools软件便能够正常运行了,不会出现加载过程中闪退的问题。
图 3 用MGLTools打开.cif后缀的蛋白(2Q1M)结构文件
PyMol的准备
PyMol:由Warren Lyford DeLano编写的基于Python编程语言的分子三维结构可视化分析软件,适用于创作高品质的小分子或是生物大分子(特别是蛋白质)的3D结构图像,比如输出受体和配体对接后的复合物3D结构。
(1)官方网址:https://pymol.org/
(2)PyMol安装步骤:①访问官方网站;②点击主页“Download now”跳转到下载页面,选择与自己电脑系统匹配的版本,单击下载;③下载完成之后回到桌面双击名为“PyMOL-3.1.8-Windows-x86_64”的安装文件;④“next”;⑤“I Agree”;⑥选“Just Me(recommended)”,点击“next”;⑦点击“Browse”选择安装位置,注意必须选择空文件夹才能执行安装程序,且最好把软件安装在D盘,点击“next”;⑧勾选第一个和最后一个默认选项即可,点击“install”;⑨添加“环境变量”,步骤参考AutoDock vina的安装,环境变量添加好之后便可以使用PyMol软件了。
图 4 用PyMol打开.cif后缀的蛋白(2Q1M)结构文件
Java环境的准备
Java:安装Java主要是为了支持LigPlot+的使用,因为LigPlot+是一款基于java界面运行的2D分子间相互作用图绘制软件。
(1)官方网址:https://www.oracle.com/cn/java
(2)Java的安装步骤:①访问官网,点击“下载Java”;②根据电脑系统选择需要的软件版本,如JDK25菜单下的windows版本的安装包/程序,“x64 Compressed Archive”为压缩包,“x64 Installer”为非压缩包的安装驱动程序,根据需要进行选择即可;③双击“x64 Installer”,点击“下一步”,选择安装地址(尽量安装到D盘),点击“下一步”开始安装。
LigPlot+的准备
LigPlot+:又称LigPlus,是一款可以绘制2D分子间相互作用图(如蛋白质与小分子配体结合模式图)的软件,在结构生物学和药物设计等领域发挥着重要作用。需要注意的是,LigPlot+仅对学术用户免费(需验证身份),下载前必须使用教育邮箱(教育邮箱一般需要通过所在学校/研究机构官网进行申请和注册)进行注册认证,完成认证后方可获得软件下载权限。
(1)官方网址:Edge浏览器搜“LigPlot+下载”
(2)LigPlot+安装步骤:①访问网址;②点击“acdemic licence”,填写个人姓名、所在单位、教育邮箱等信息,填写完成后点击“accept”即可;③认证通过后回到最初的界面输入邮箱地址和密码(登陆填写的教育邮箱查看),点击“download”即可开始下载;④下载好之后解压文件夹到D盘,路径中最好不要有中文;⑤打卡解压后的文件夹,双击文件“LigPlus”,后缀为.jar,选择Java作为打开应用,随后会加载出来一个提示弹窗,点击“确定”;⑥进入路径和目录设置界面,这个地方可以将PyMol的.exe路径复制到相应位置(如D:\PyMol\PyMOLWin.exe),实现软件的“串联”,即在LigPlot+中调用PyMol实现2D互作图的3D显示;⑦设置好后点击“save”即可开始使用。
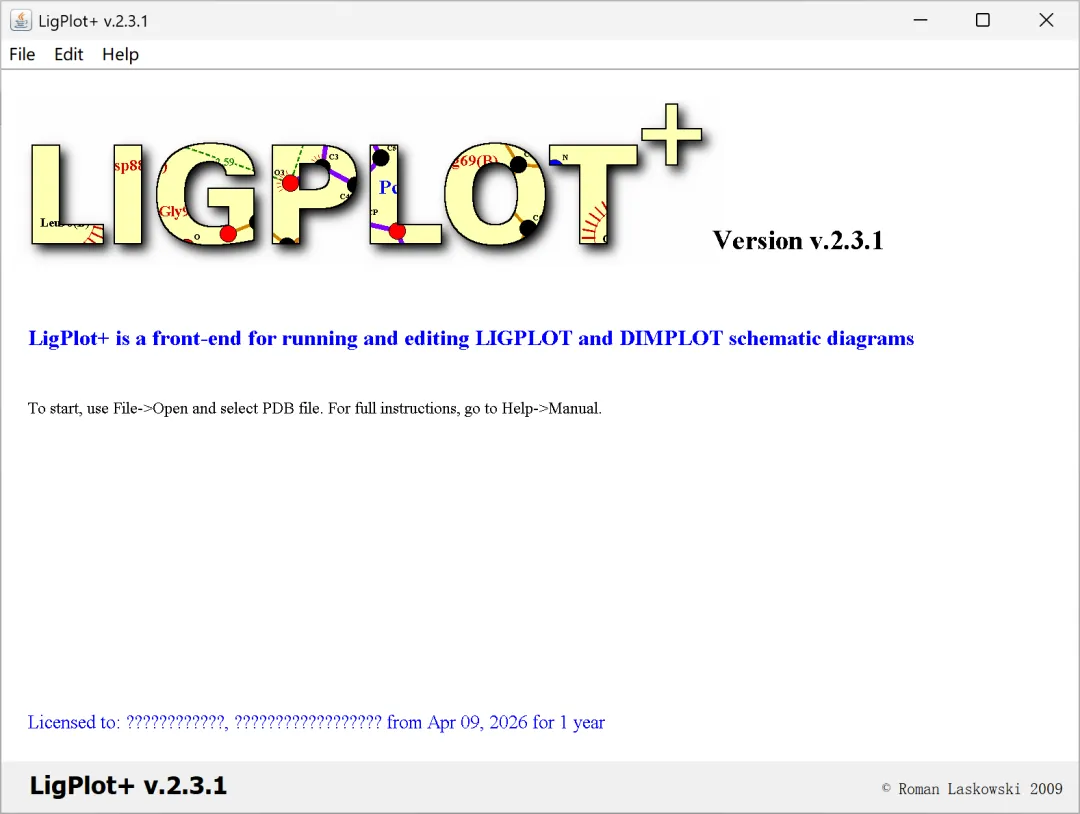
图 5 LigPlot+安装成功后正常运行的操作界面
注:如您需要安装该软件,但是又无法注册教育邮箱的,可以后台私信借用教育邮箱下载该软件。
Open Babel的准备
Open Babel:是一个开源的化学工具和库,主要用于分子数据的转换、处理和可视化。它支持许多分子文件格式,可以将不同的分子数据格式之间进行转换,也可以进行各种化学计算。Open Babel在分子对接中的应用主要是进行小分子配体化合物结构文件的格式转换。
(1)官方网址:https://openbabel.org/index.html
(2)Open Babel安装步骤:①访问官网→“User Guide”→“Install Open Bable”→“GitHub”→选择“OpenBabel-3.1.1-x64.exe”版本进行下载;②双击启动安装驱动程序→“next”→“I Agree”→“Browse”选择保存地址,尽量存在D盘下→“next”→“install”→“Finish”。
图 6 Open Bable安装成功后正常运行的操作界面
ChemDraw的准备
ChemDraw:ChemBioOffice核心工具之一,提供一套完整易用的绘图解决方案,包括绘制化学结构及反应式,并且可以获得相应的属性数据、系统命名及光谱数据等,由美国Revvity公司出品。在分子对接中的应用主要是绘制在PubChem中无法查到结构信息的小分子配体化合物的3D结构,具体利用的是Chem3D配套组件。
(1)官方网址:https://www.chemdraw.com.cn/
(2)ChemDraw安装步骤:ChemDraw是一款非开源的付费软件,如果不经常使用可以申请一段时间的免费试用。如果需要长期使用,除个人单独购买以外,还可以查询所在学校或单位官方网站(一般是网信中心或专用的软件平台),看是否有统一购买正版软件授权。
注:网上有很多可获取的软件资源(自行查找)。
Adobe illustrator的准备
Adobe illustrator:由Adobe公司开发的矢量图形处理软件,集成文字处理、上色等丰富的功能,操作简单,功能强大。在分子对接中的应用主要是图像的拼接处理。
(1)官方网址:https://www.adobe.com/cn/
(2)Adobe illustrator安装步骤:Adobe illustrator同样是一款非开源的付费软件,可以个人购买或查询所在学校/单位官方网站(一般是网信中心或专用的软件平台),看是否有统一购买正版软件授权。
注:如果实在找不到软件资源,用Powerpoint、Word等软件也可以完成图片的拼接,但是个人还是更推荐使用Ai,因为专业图形软件的可操作性和便捷性是远远大于普通办公软件的。
数据库的准备
UniProt(Universal Protein)数据库
UniProt(Universal Protein)数据库:一个提供蛋白质序列与功能信息的全球化综合性数据库,于2002年由Swiss-Prot、TrEMBL和PIR三大数据库组织联合创建,提供覆盖范围广泛的蛋白质序列、各种变体、功能特征、细胞定位、相互作用等信息。UniProt在分子对接中的应用主要是查找目标受体蛋白的ID(Entry)。
官方网址:https://www.uniprot.org/
RCSB PDB数据库
RCSB PDB数据库:全布鲁克海文蛋白质数据库(The Brookhaven Protein Data Bank,简称PDB)是由美国布鲁克海文国家实验室于1971年创建的国际性生物大分子三维结构数据库。该数据库由结构生物信息学研究合作组织(RCSB)维护,收录通过X射线单晶衍射、核磁共振和电子衍射等实验手段测定的蛋白质、核酸及糖类等生物大分子结构数据,内容包括原子坐标、晶体结构因数、NMR实验数据及相关参考文献,是wwPDB世界蛋白质数据库的一部分。在分子对接中的应用是根据UniProt中获得的目标受体蛋白的ID(Entry)查找目标受体蛋白的3D结构文件。
官方网站:https://www.rcsb.org/
PubChem数据库
PubChem数据库:是一种化学模组的数据库,该数据库由美国国家生物信息中心开发,涵盖化学信息学、化学生物学、药物化学和药物发现等诸多领域的重要化学信息资源,被广泛用于药物虚拟筛选、药物再利用、化合物毒性预测、药物副作用预测和代谢物鉴定等研究。在分子对接中的应用主要是获取小分子配体化合物的3D结构文件。
官方网址:https://pubchem.ncbi.nlm.nih.gov/
受体和配体的准备及处理
受体分子的准备与处理
受体分子的准备:①以TNF(肿瘤坏死因子)为例,首先在Uniprot数据库中搜索栏输入TNF,点击“search”,左侧筛选栏设置筛选条件如“Reviewed(已被验证)”“Human(人源)”,找到需要的蛋白如“TNFSF18”并复制它的“Entry”-Q9UNG2;②打开RCSB PDB数据库,粘贴“Q9UNG2”进行搜索,查找最需要的蛋白结构如PDB ID为“7KHD”的蛋白结构,打开详情页,点击“download files”,选择下载格式,一般都选择“PDBx/mmCIF Format”格式,文件后缀为.cif,下载完成后将该文件保存到安装AutoDock vina的文件夹路径之下,如前面教程中提到的“D:\AUTODOCK”。
注:后续涉及到的所有文件最好都放在该路径中,目的是方便查找和处理文件,专业一点说就是将该文件夹视为“工作目录”,后面涉及到软件的具体使用时会提到。
受体分子预处理:①用PyMol打开打开下载的““7KHD”蛋白结构PDB文件,后缀为.cif;②删除水分子(H2O),即图像中一个个红色的小+号,具体步骤为点击并打开右侧All条目“ASHLC”中的“A”选项卡,选择“remove waters”;③删除金属离子,结构中的绿色球体便是金属离子,具体步骤为点击操作界面右下角的“SEQ”打开蛋白质序列,单击选中上方显示出来的一行绿色的蛋白序列中所有金属离子,点击打开右侧(sele)的“A”选项卡,选择“remove atoms”,也可以单击右键选择“remove”进行删除;④删除配体,步骤和金属离子的删除相同,点击操作界面右下角的“SEQ”打开蛋白质序列,滑动滑块到序列最后,单击选中所有配体(三个字母),点击打开右侧(sele)的“A”选项卡,选择“remove atoms”,当然也可以直接在图中单击选中配体进行删除,但是这样比较麻烦,且容易漏删,所有不太建议如此操作;⑤导出处理好的蛋白结构文件,点击“File”→“Export structure”→“Export molecule”→“Save”,选择保存地址,命名为“7KHD_ST”并选择后缀为.pdb/.pdb.gz的PDB文件进行保存。
注:下载的7KHD蛋白结构文件中似乎不含水分子和金属离子信息,但是最好不要跳过删除步骤,仅通过肉眼观察容易忽略掉被其他结构遮住的应该去除的分子/离子。
配体分子的准备与处理
配体分子的准备:小分子配体化合物使用RCSB PDB数据库中明确给出的7KHD蛋白配体C8H15NO6(N-Acetyl-D-Galactosamine)①首先打开PubChem数据库,搜索“N-Acetyl-D-Galactosamine”,一般选取BEST COMPOUND MATCH栏目下的化合物,点击化合物结构图或名称可进入化合物结构信息详情页;②点击structure栏目的3D图片可跳转至3D结构展示界面,点击“download coordinates”,选则SDF格式的3D结构文件,点击“Save”进行下载,保存到工作目录中,修改名字为“NADG”。
配体分子(小分子化合物)预处理:配体分子一般不需要像受体(蛋白)一样进行处理,但是需要进行SDF结构文件的格式转换,将其修改为PDB格式。具体的处理步骤为打开OpenBabel,左侧操作栏INPUT FORMAT下方下拉选项选择需要进行格式转换的文件后缀类型如“sdf-MDL MOL format”→点击C:\WINDOWS下方空格右侧的小矩形打开“Choose input file”窗格后选择需要进行格式转换的小分子配体SDF文件→右侧操作栏OUTPUT FORMAT下方下拉选项选择要转换为的PDB文件格式→取消勾选“Output below only(no output file)”,然后点击output file下方空格右侧的小矩形打开“Choose out put file”窗格选择转换格式后的文件保存地址并命名,如“NADG_PT”→点击中间操作栏的“Do conversion”完成转换(快捷键Alt+C)。
注:OpenBable的操作界面有一点显示问题,部分文本内容显示不完整,可以将鼠标光标在未显示完整的文本或操作键上多停留一会儿以显示提示词。
开始分子对接
在准备好受体和配体分子的结构信息文件后,便可以开始做进一步的处理并进行分子对接了。
受体分子文件的导入和处理:①打开AutoDockTools,即MGLTools,点击“File”→“Preferences”→“set”→“startup Directory”位置输入AutoDock vina所在路径D:\AUTODOCK作为工作目录(如下载受体分子结构文件时所述,主要是为了方便后续查找分子对接所涉及的一系列文件,同时保证对接程序能够正常地运行)→“Make default”;②点击“Grid”→“Macromolecule”→“Open”→更改文件后缀为.pdb,选择经过去水、去金属离子、去配体处理的受体蛋白结构文件,如“7KHD_ST.pdb”,打开后不用另存为,直接取消即可,跳出的警告和提示窗则均点“确定”;③加氢:点击“Edit”→“Hydrogens”→“Add”→“ok”;④再次点击“Grid”→“Macromolecule”→“choose”→选中“7KHD_ST”后点击“Select Molecule”将加完氢的分子选为受体,此时会弹出一个提示窗格,点击“确定”将受体分子文件另存为.pdbqt后缀的新文件,保存到工作目录,这样即完成了受体分子的导入和处理。这时候再将导入的“7KHD_ST”受体文件从软件列表中删除(左侧导航栏单击选中“7KHD_ST”之后打开右键菜单“delete”)。
注:工作目录设置成功后,之后再使用AutoDockTools时就不用再重复设置了。另外,此处提到的加氢是化学结构上的真实物理变化,对比加氢前的结构并非简单的显示与不显示的区别。之所以要加氢是因为蛋白质的晶体结构大多都是利用X射线衍射分析测得的,而由于氢只有1个电子,电子云密度极低,难以利用X射线衍射进行检测和观测。所以实际上从RCSB PDB数据库中下载的蛋白结构本身就是不完整的,一般只记录了C/N/O/S等骨架结构,而不含H的信息,这里所谓的“加氢”操作相当于是为了得到完整的蛋白结构。
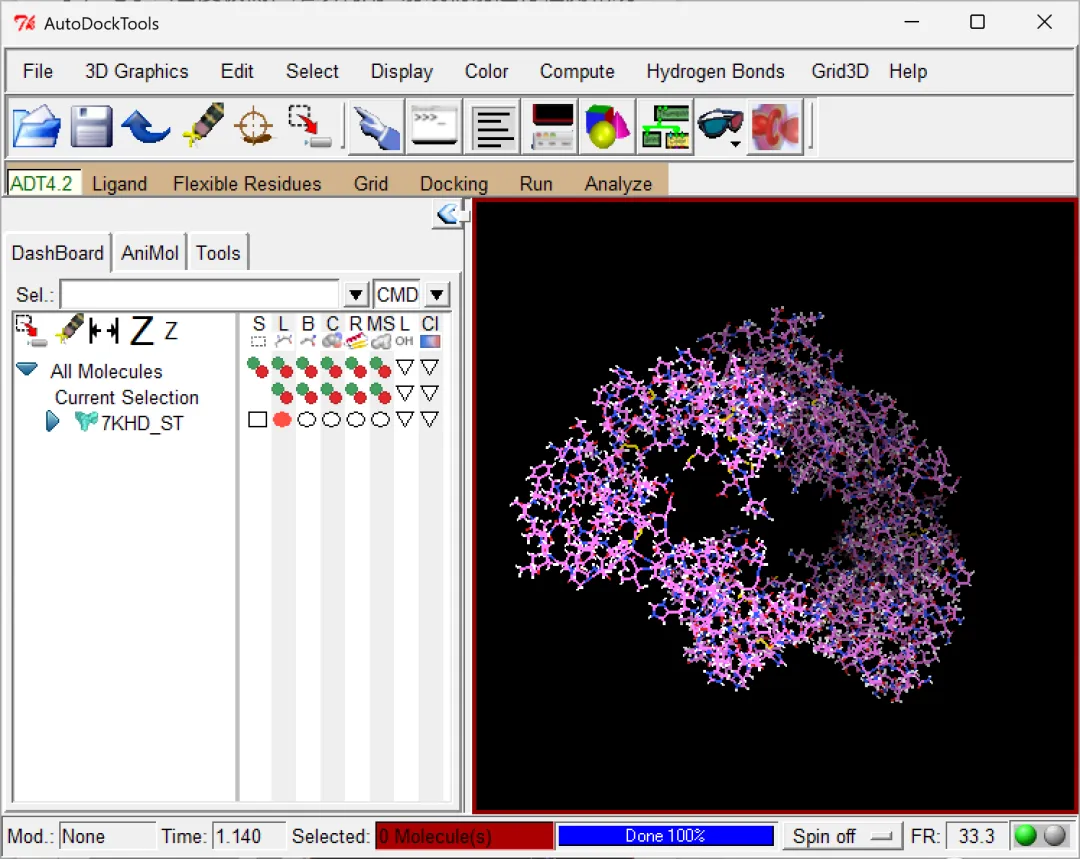
图 7 “加氢”后的7KHD蛋白结构
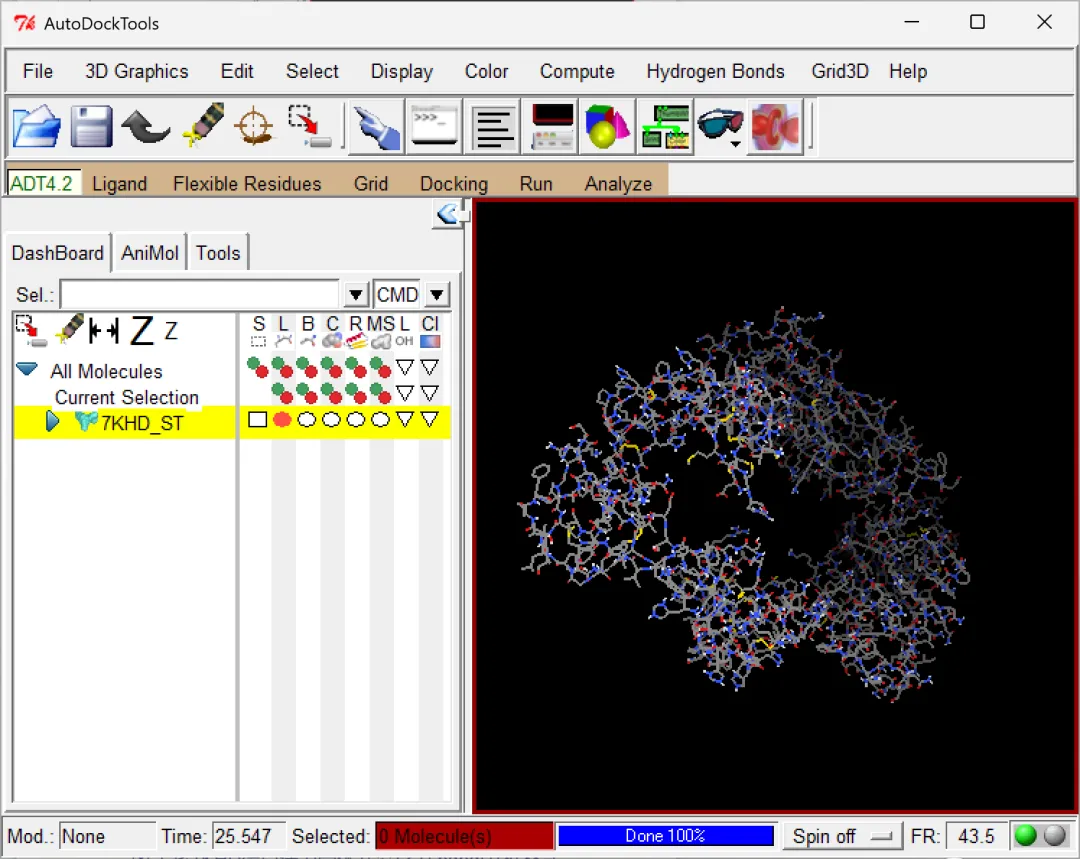
图 8 将7KHD蛋白选为受体并另存为.pdbqt文件
配体分子文件的导入和处理:同样使用AutoDockTools进行处理①点击“Ligand”→“input”→“open”→更改文件后缀为.pdb,选择经过格式转换的.pdb后缀的小分子配体化合物结构文件如“NADG_PT.pdb”,此时会弹出一个提示框,直接点“确定”即可;②小分子配体化学物的结构一般无需进行任何处理,直接点击“Grid”→“Set Map Types”→“Choose Ligand”,选中配体文件名称后点击“Select Ligand”将该分子选为配体,这时候会弹出一个提示框,直接点击“确定”即可;③确定扭转树的根原子(绿球),点击“Ligand”→“Torsion Tree”→“Detect Root”(配体中通常含有多个可旋转单键,点击Detect Root会选定一个“根”原子,以此为起点构建分子的扭转树-torsion tree,用于描述构象空间);④可旋转键计数,即标记可以旋转的单键(数根绿色填充的化学键),点击“Ligand”→“Torsion Tree”→“Choose Torsions”,查看完之后点击“Done”;⑤点击“Ligand”→“Output”→“Save as PDBQT”,同样再将导入的配体文件从软件列表中删除(左侧导航栏单击选中“NADG_PT”打开右键菜单“delete”)。
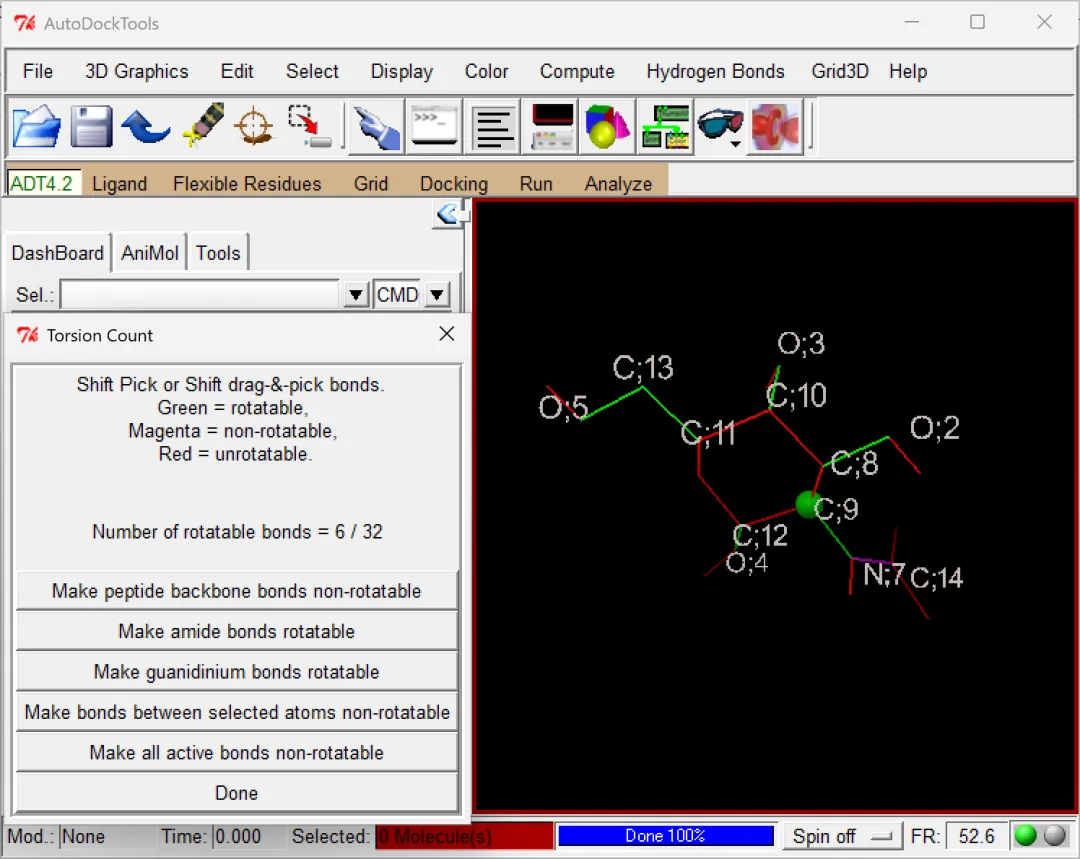
图 9 标记了根原子和可旋转单键的小分子配体结构
开始受体和配体分子的对接:使用AutoDockTools完成受体配体的对接①点击“Grid”→“Macromolecule”→“Open”,导入.pdbqt后缀的受体蛋白结构文件→“yes”→“确定”;②点击“Grid”→“Set Map Types”→“Open Ligand”,导入.pdbqt后缀的小分子配体结构文件→“确定”;③设置对接(网格)盒子的大小:点击“Grid”→“Grid Box”,在“Grid options”窗格中左右拉动各个齿轮以调节对接盒子的大小和位置,尽量使得整个受体蛋白结构位于对接盒子中→点击“Grid options”窗格的“File”→“Close Saving Current”;④导出对接文件:“Docking”→“Output”→“vina config(config.txt)”,跳出的窗格点击点击“show options”可以设置“对接次数modes(默认对接9次)”等参数→“Save”,这时候会在最初设置的工作目录D:\AUTODOCK输出一个.txt格式的对接文件;⑤开始对接:注意开始对接前,对接文件、受体配体结构文件和vina的驱动文件需放在同一个文件夹内。点击“Run”,选择“Run AutoDock Vina”→在“Config Filename”输入框中输入或点击“Browse”打开前一步导出的对接文件“config.txt”(正常情况下一般已经自动选择了该文件),点击“Launch”正式开始对接,接着会弹出来一个“Autodock Process Manager”窗口,说明Autodock Vina驱动程序被调动且正常运行,这时候已经在执行对接程序了,不要关闭该窗口,也不要进行任何操作,此时可以在命令窗口查看对接进度,当对接完成后“Autodock Process Manager”窗口会自动关闭;⑥窗口关闭后,在命令窗口可以查看输出的分子对接数据表格,其中包含多次对接都结果,主要提供Affinity(结合能)大小等关键信息,同时还会在工作目录输出一个名称后带有“_out”的.pdbqt后缀的分子对接结果文件,如“NADG_PT_out.bdpqt”。
注:调整网格/对接盒子时,先调节xyz三个轴的数值,如果调节到最大值还无法包住整个蛋白结构,则可以调节“Spacing”的大小。注意不要将小分子配体包入对接盒子,显示界面中的受体蛋白、配体分子结构都是可以拖动的,以移动小分子配体的结构为例,点击操作界面上方一排图形中的“DejaVu GUI(球+圆锥+正方体,鼠标光标在上面多停留一会儿会显示名称)”→取消勾选“mouse transforms apply to “root” object only”→点击“+”打开“root”列表→单击选中小分子配体结构文件名如“NADG_PT”,在右侧结构图中按住鼠标右键可以对小分子配体结构进行单独拖动,鼠标左键进行单独旋转,选中受体蛋白结构文件名则可以单独拖动或选择受体蛋白的结构。如果要调整整个画面,记得重新勾选“mouse transforms apply to “root” object only”或者直接选中“root”。
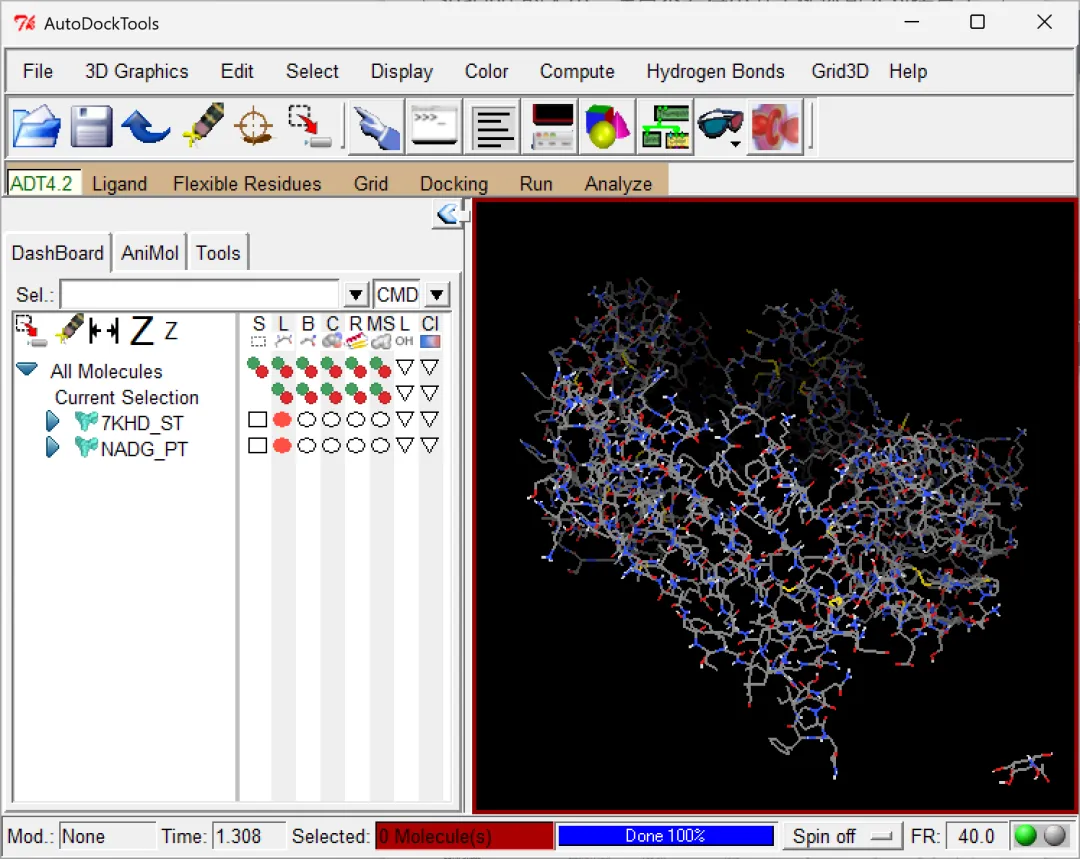
图 10 在AutoDockTools中导入受体和配体的.pdbqt文件
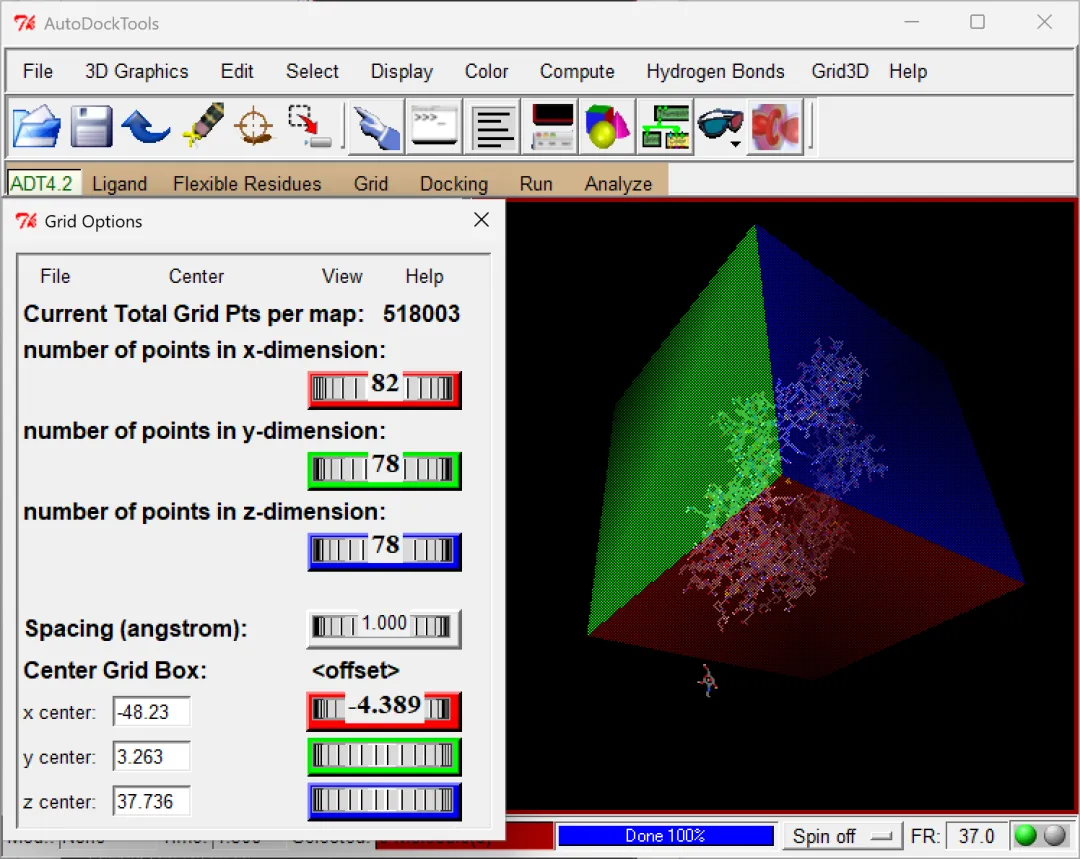
图 11 调整参数使整个受体蛋白结构位于对接盒子中
图 12 执行对接程序图中文件必须在同一工作目录/文件夹
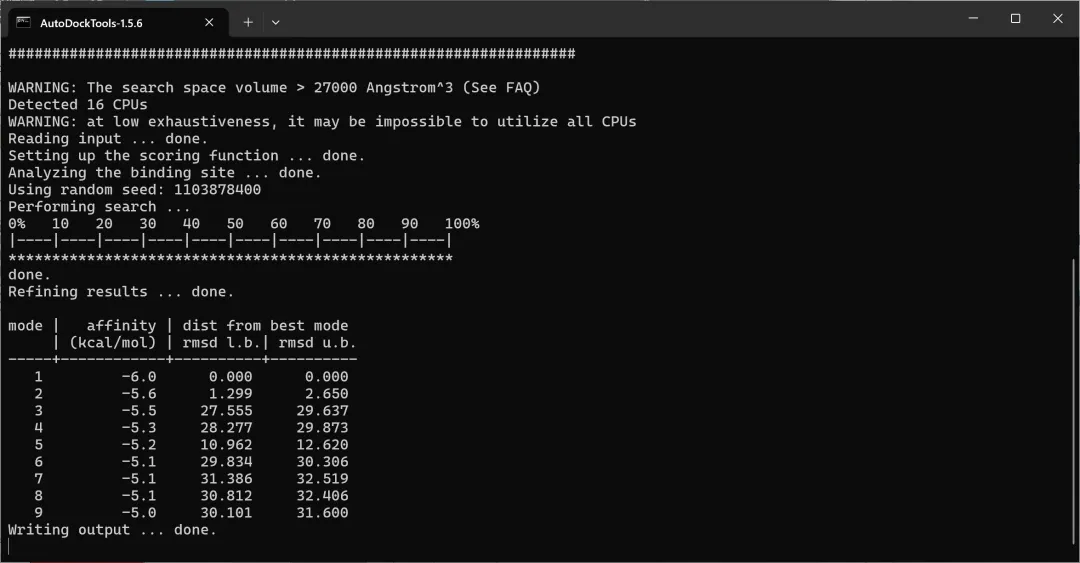
图 13 在命令窗口可查看分子对接的百分比进度和结果数据
注:一定要注意区分结合亲和力与结合能,前者的大小与分子间结合的稳定性成正比,而后者的大小则与分子间结合的稳定性成反比。在AutoDock vina输出的分子对接结果表中,虽然表头显示的是affinity(kcal/mol),但其含义为结合能而非亲和力,这一点需要注意。其中第三列的“dist from rmsb l.b.”全称为distance from RMSD lowest bound,代表意指当前构象与结合能最低(即最优)构象之间的RMSD(均方根偏差)下限值,反映该构象相对于最优构象的空间偏移程度(第一个即为最优构象,所以返回结果为0)。而第四列的“best mode rmsb u.b.”全称为best mode RMSD upper bound,是指当前构象与最优构象之间的均方根偏差上限,同样用于衡量该构象与最优构象的结构差异程度。
对接结果分析及可视化
分子对接结果分析
受体与配体之间的互作分析:利用AutoDockTools完成,①首先将前面导入的受体、配体文件删除:点击“Edit”→“delete”→“Delete All Molecules”→“CONTINUE”;②开始分析:点击“Analyze”→“Dockings”→“Open Autodock vina result”,选择输出的.pdbqt格式分子对接结果文件→“打开”→“ok”→“确定”→点击键盘左右键可以切换每次对接的不同构象(为按照结合能从小到大的顺序进行的构象排序,也就是说显示的第一个构象即为结合能最小的构象);③打开受体的结构文件:“Analyze”→“Macromolecule”→“Open”,选择受体蛋白.pdbqt格式的文件→“打开”→在左侧分别点击“SLBCR…”等选项下的图形可(圆形、三角形、正方形)调整受体和配体分子结构的显示格式等(鼠标光标在图形上多停留一会儿能够显示具体的功能描述)。同时点击键盘左右键可以查看每次不同构象(结合能不同)的小分子配体与受体蛋白的结合情况;④查看分子间的互作情况:首先点击“Analyze”→“Docking”→“Show interactions”,在“Binding Site Interactions display options”窗口勾选“display pi-pi interactions”可以查看是否有π-π相互作用,勾选“display pi-cation interactions”可以查看是否有π-阳离子相互作用。以示例受体配体为例,结果显示两种互作都不存在,但从“Show Hydrogen Bonds as Spheres”窗格看到显示“2 Atoms in hBonds”,具体为“NADG_PT_out::UNL1:H 1(viseble)”和“7KHD_ST:A:ASN80:HD22 0(not viseble)”,表示小分子配体NADGD的第1号UNL残基上的氢原子与受体7KHD蛋白A链上第80号天冬酰胺(ASN)残基的HD22号氢原子之间生成了一个氢键;⑤点击键盘左右键同样可以进行不同构象小分子配体和受体蛋白结合情况的切换,重复前述分析过程即可查看和分析其他构象小分子配体与受体蛋白的结合情况。
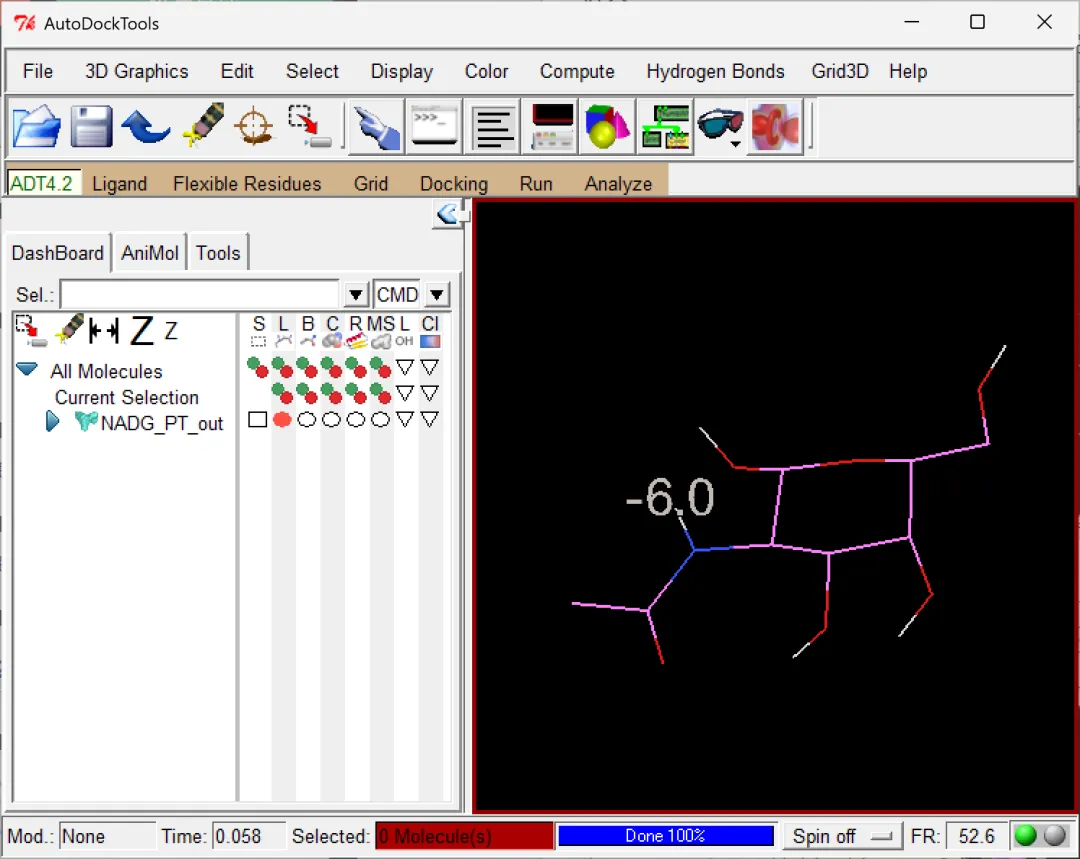
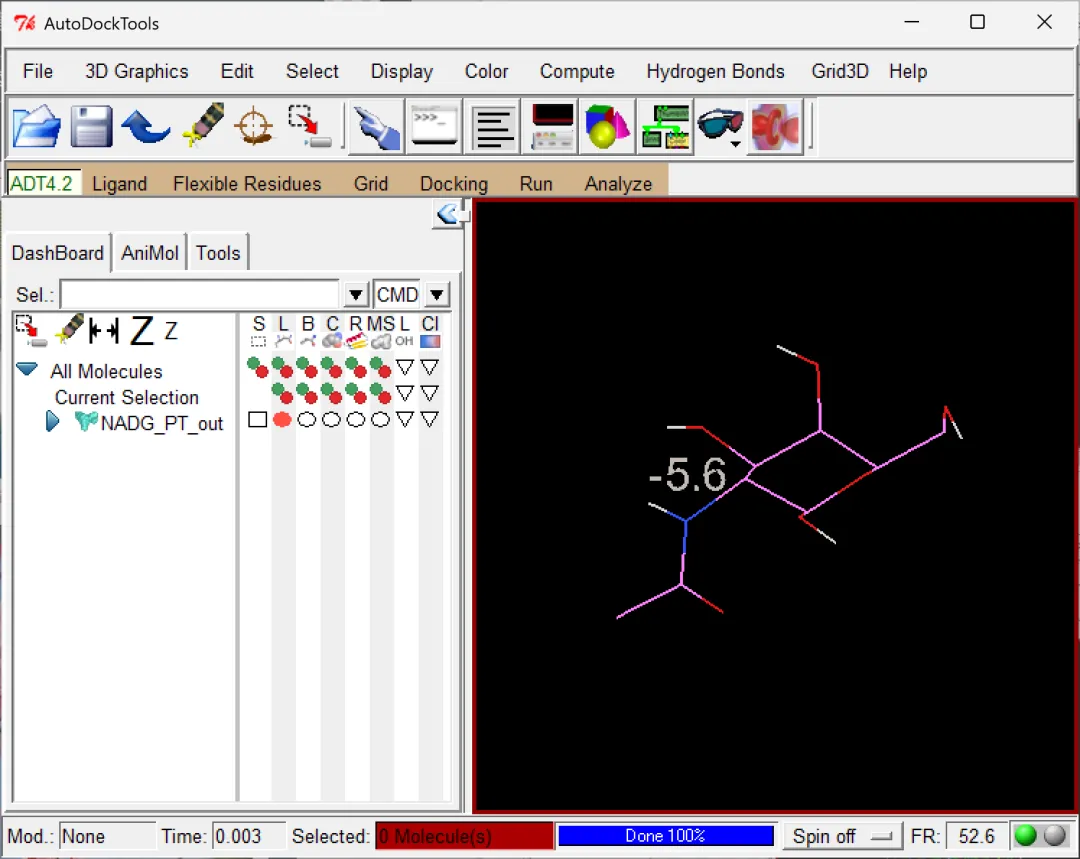
图 14 结合能从小到大排列的不同构象配体(左右滑动查看)
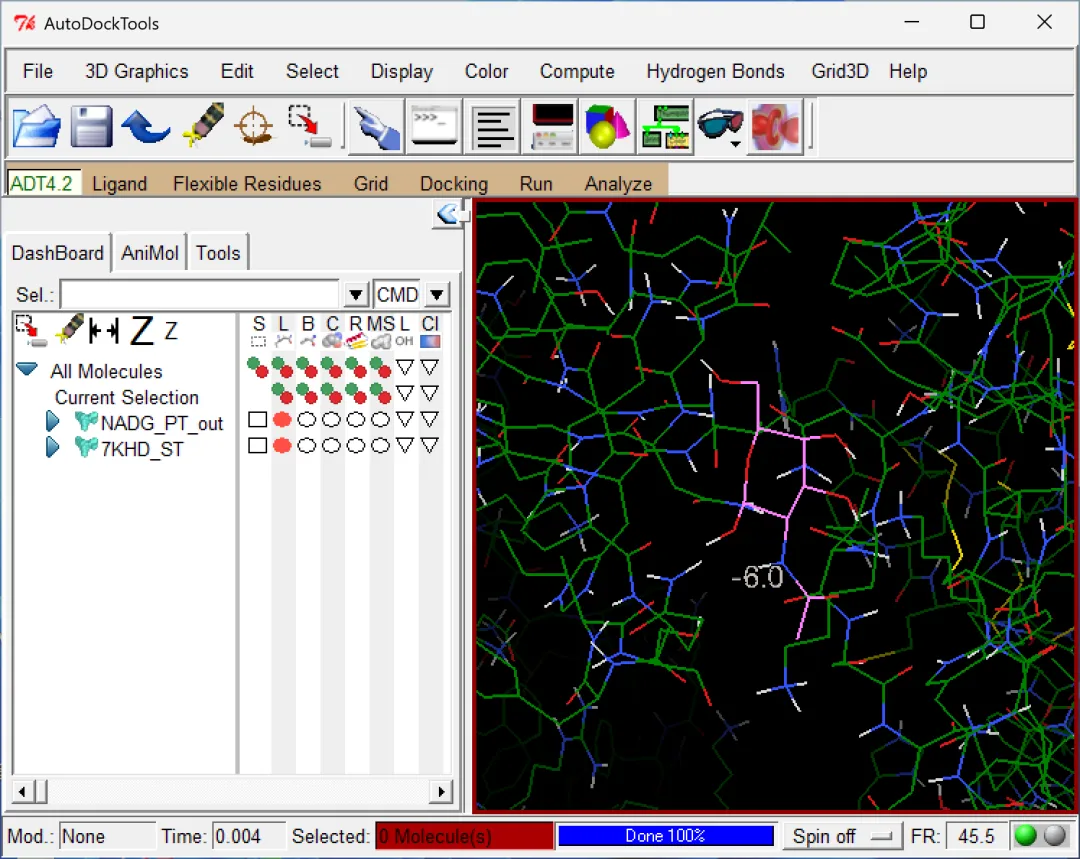
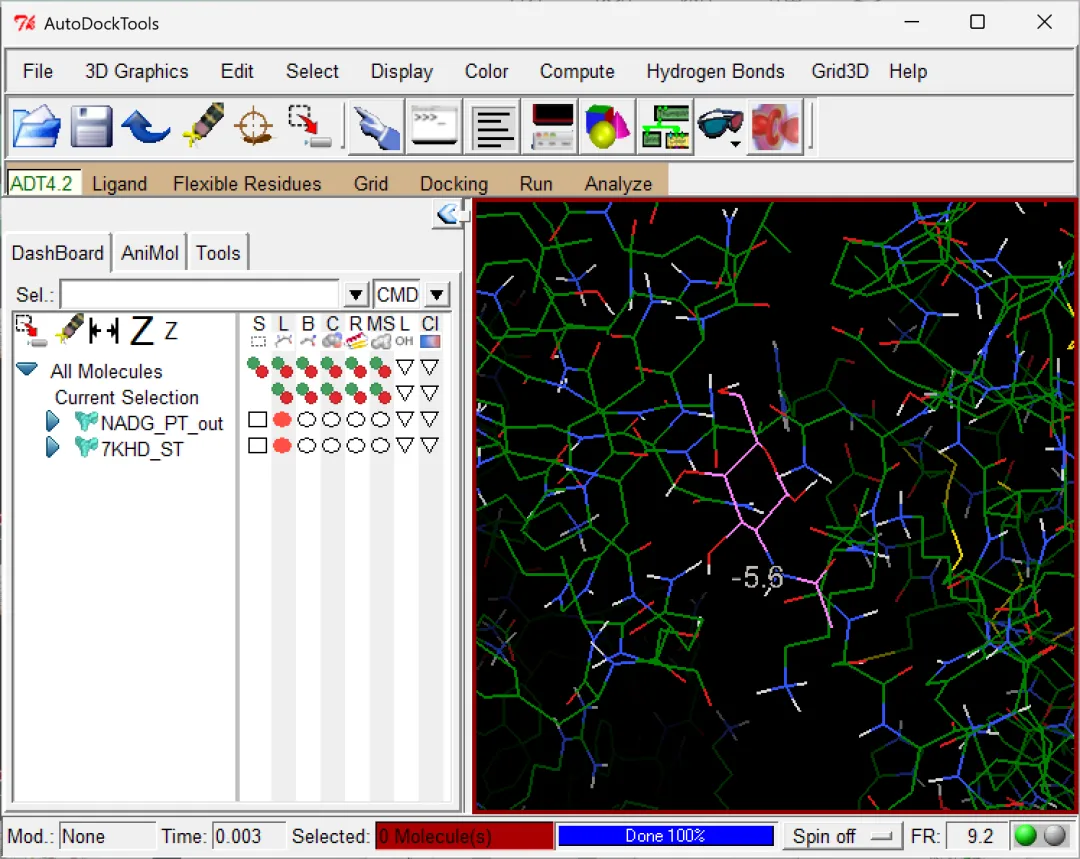
图 15 不同构象配体与受体的结合情况(左右滑动查看示例)
其实从这里就能看出来使用的是“半柔性对接方法”
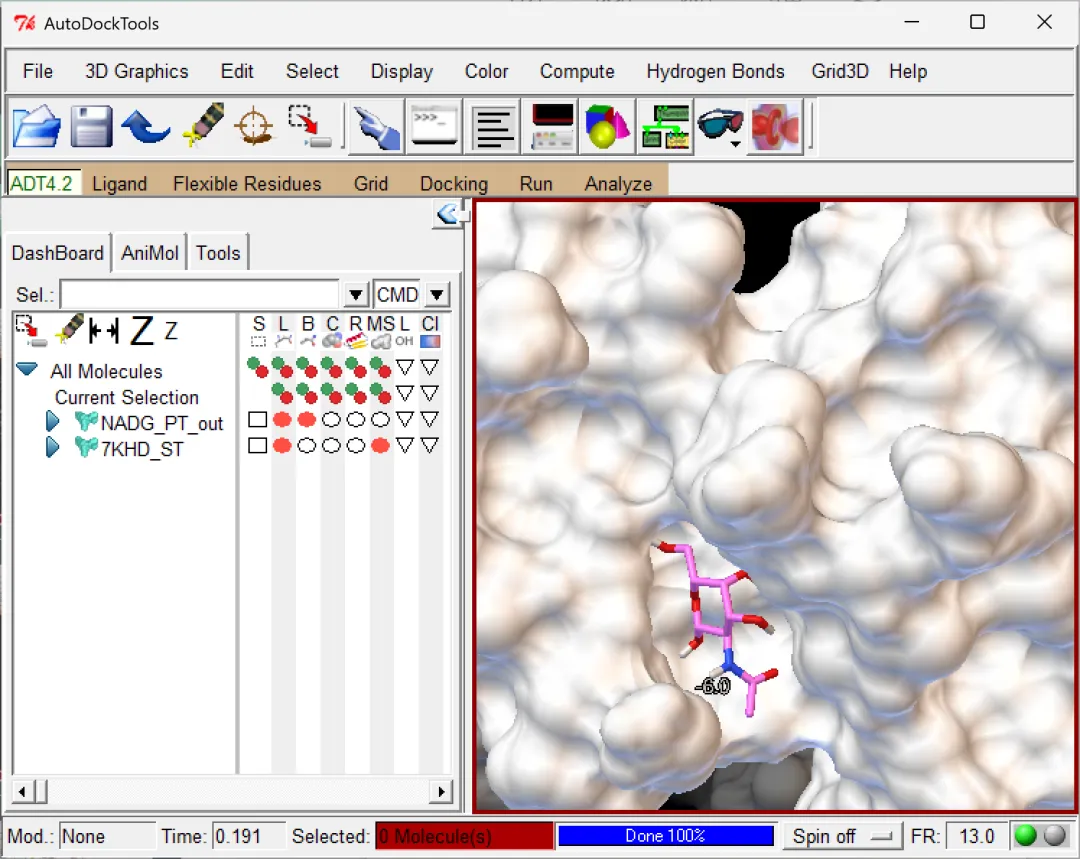
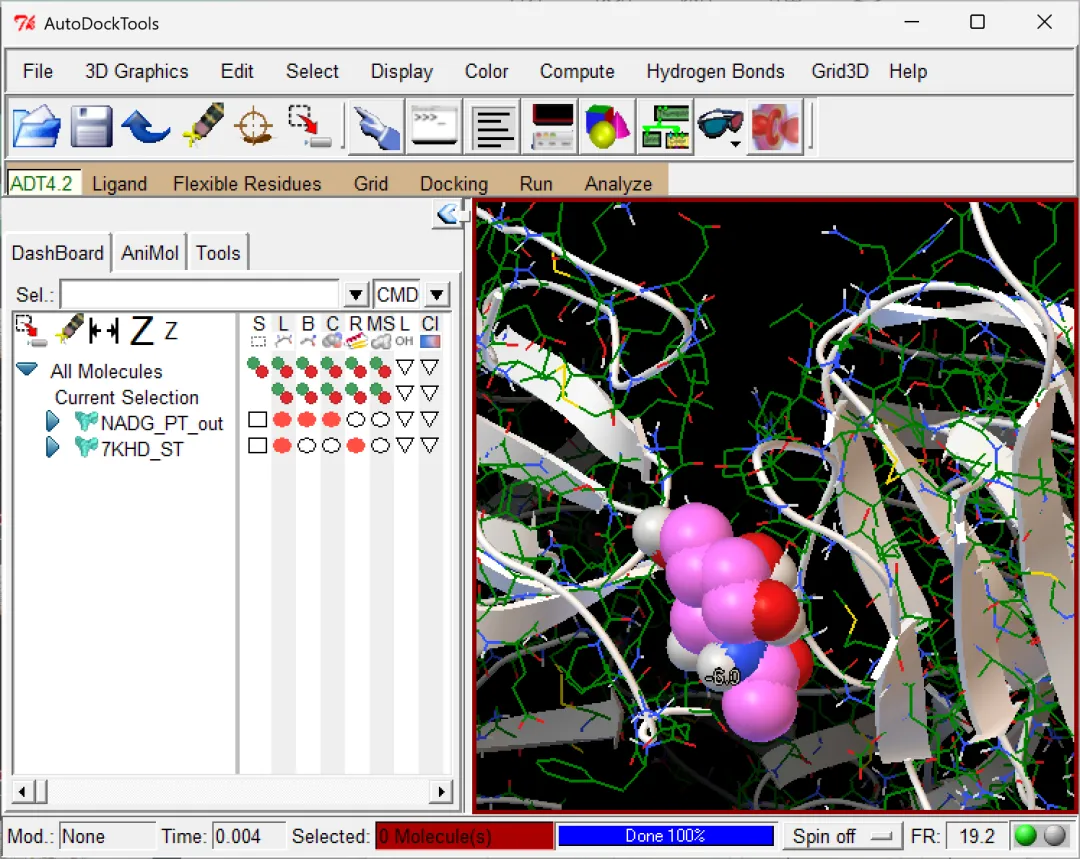
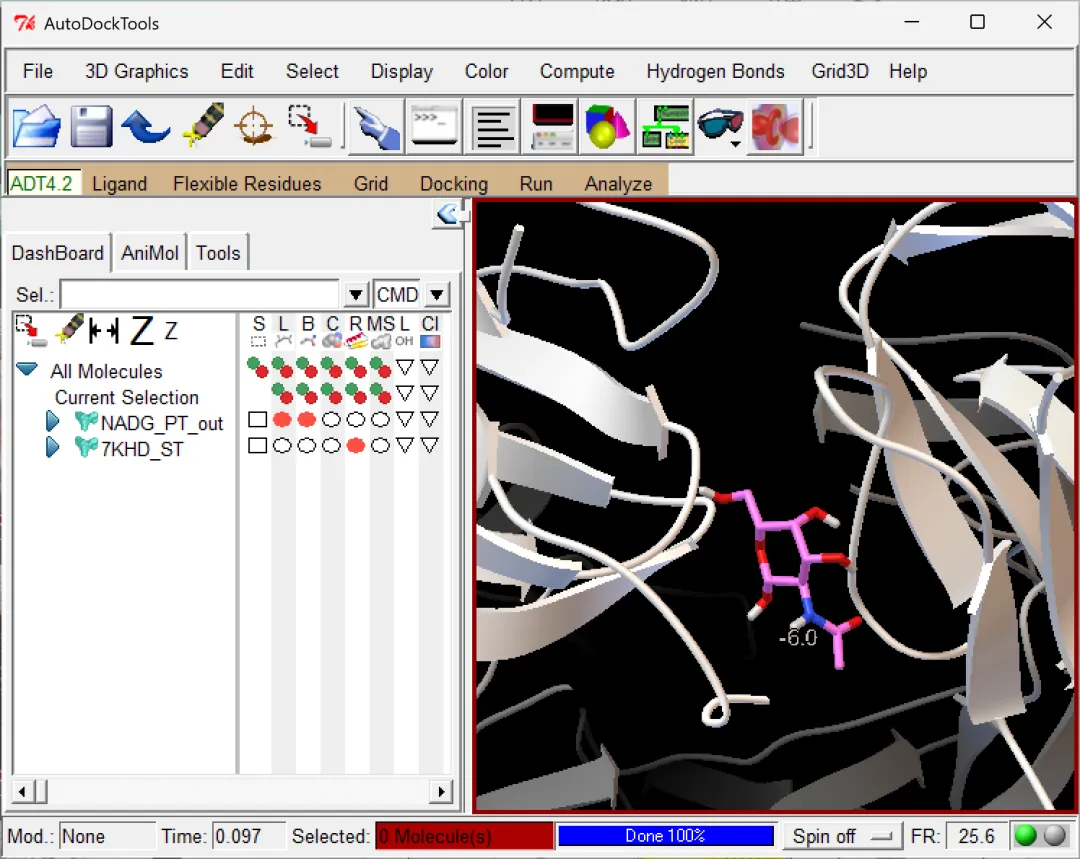
图 16 显示方式可以根据喜好进行调整(左右滑动查看示例)
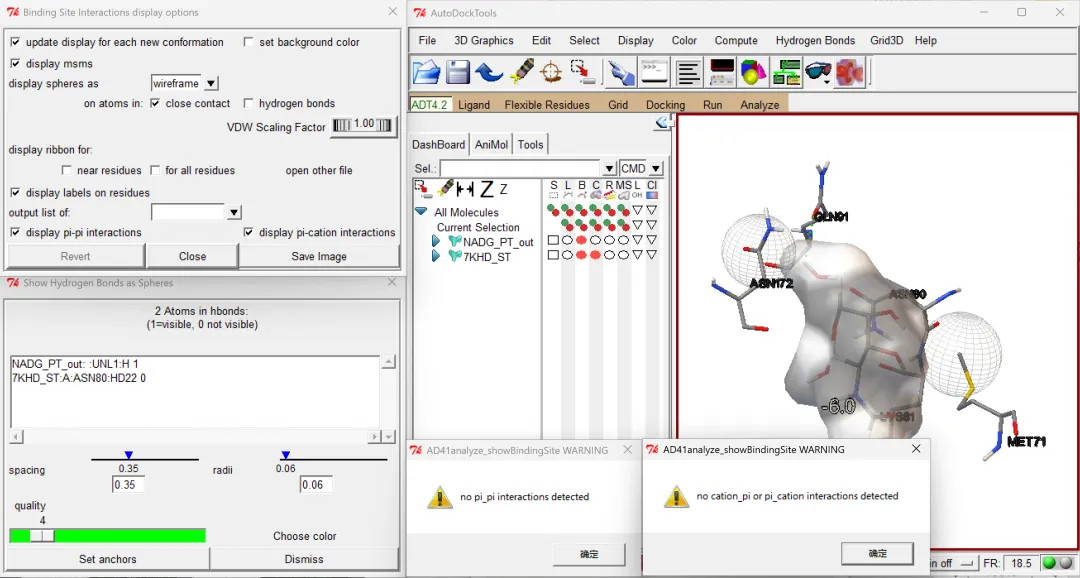
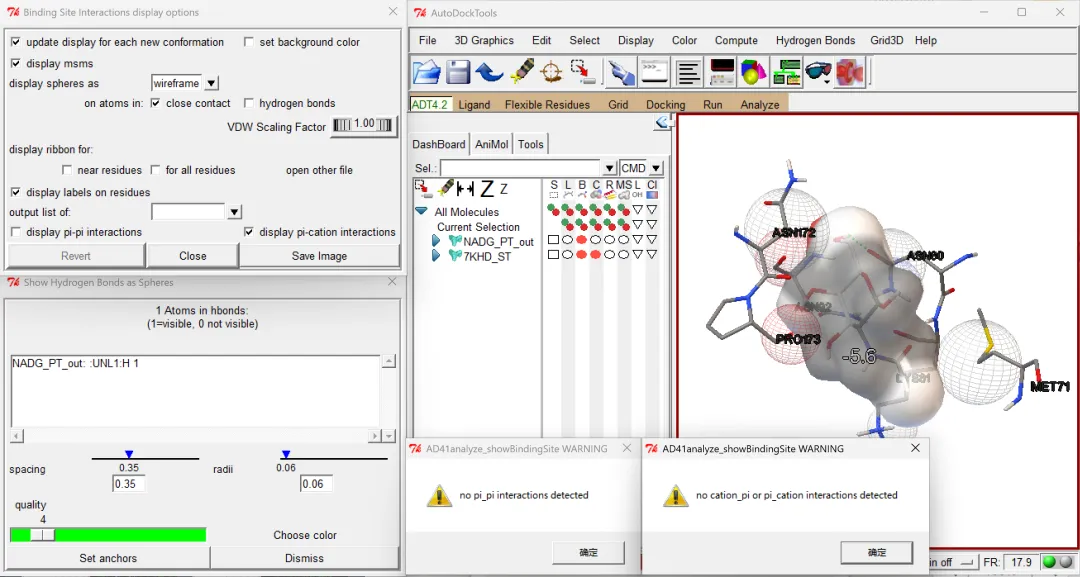
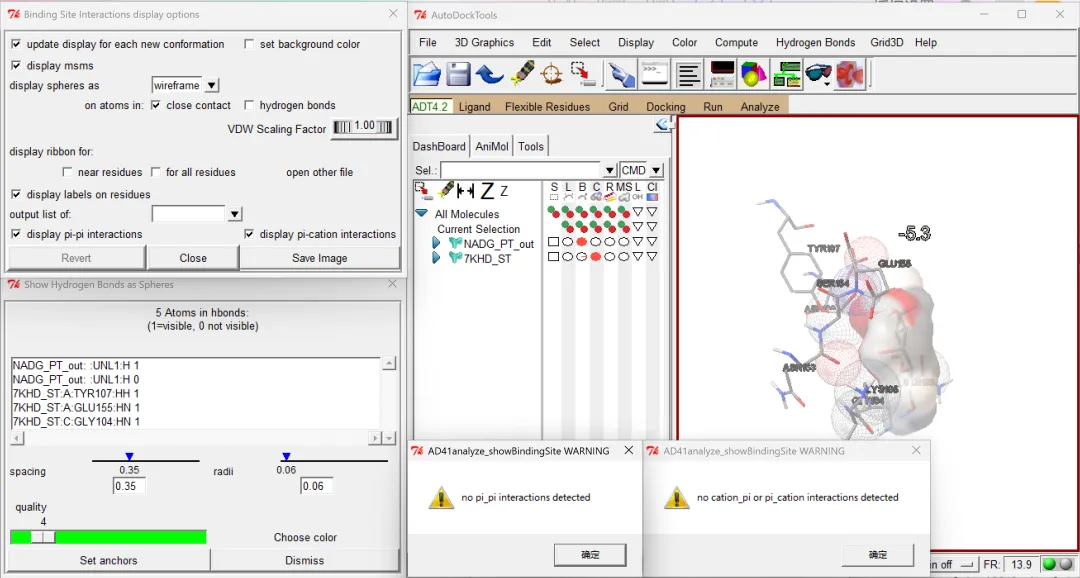
图 17 分子间π-π/π-cation/氢键生成等互作分析
(左右滑动查看示例)
输出受体和小分子配体对接后的复合物:用AutoDock Tools结合PyMol实现该功能①打开AutoDock Tools,点击“Analyze”→“Docking”→“Open Autodock vina result”,选择输出的.pdbqt后缀且名称以“_out”结尾的分子对接结果文件→“打开”→“ok”→“确定”;②点击“File”→“Save”→“Write PDB”将当前页面内的小分子配体结构(以结合能最低的构象为例)单独输出为一个.pdb格式的文件(原始.pdbqt分子对接结果文件包含所有构象的小分子配体结构信息,但我们只需要一个最佳构象的信息)→“ok”,文件同样会被保存到工作目录(文件夹)中;③打开PyMol,点击“File”→“open”,选择刚才输出的(结合能最低的)小分子配体结构.pdb文件→“打开”,同样的步骤再打开受体蛋白7KHD的.pdb文件“7KHD_ST.pdb”;④然后点击“File”→“Export structure”→“Export Molecule”→“PDB options”→“Save”,将文件命名为“FHW”,选择后缀为.pdb/.pdb.gz的PDB文件格式并将其保存到工作目录下→打开All的”A”选项卡,点击“delete everything”;⑤点击“File”→“Open”→选择刚才输出的“FHW.pdb”文件;⑥隐藏小分子配体结构中的二级键及删除氢原子:直接在结构图中点击选中小分子配体的整个结构,打开右侧(sele)的“H”选项卡,点击“valence”,继续选中小分子配体的结构,打开右侧(sele)的H选项卡,点击“Hydrogens”→“all”;⑦再次选中小分子配体的结构,打开右侧(sele)的A选项卡→“find”→“polar contacts”→“to other atoms in object”显示受体蛋白和小分子配体之间形成的氢键(黄色虚线标注)。
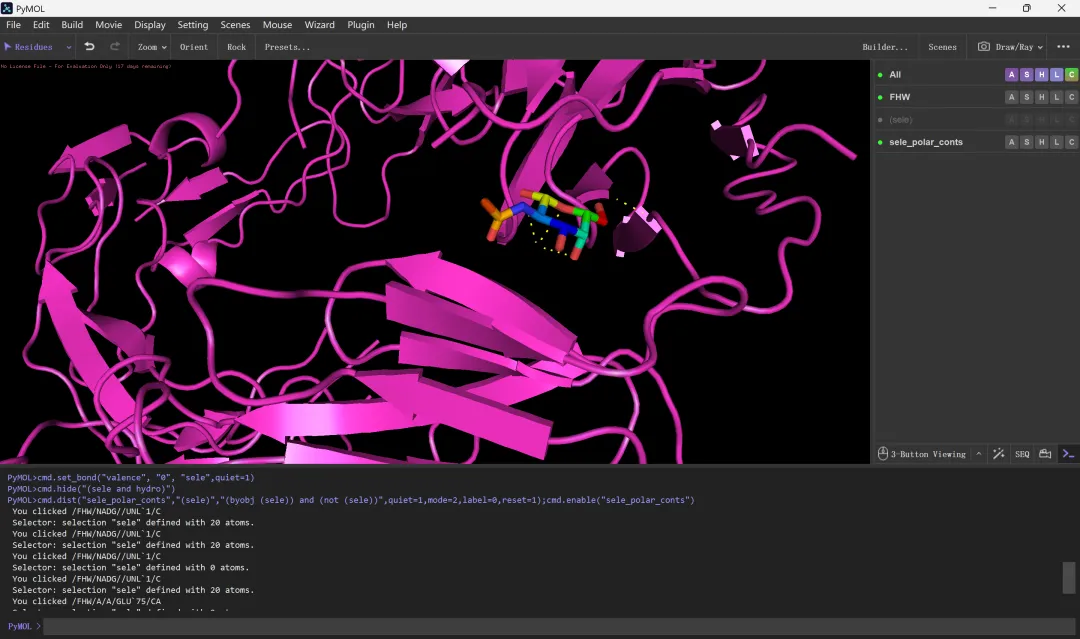
图 18 黄色虚线即为受体和配体之间生成的氢键(共4根氢键)
注:单击选中受体或配体的结构可以调整颜色等显示格式,以调整小分子配体的颜色为例,具体步骤为:选中小分子配体→单击右键→color→选择自己钟意的配色如spectrum-rainbow(elem c)。
分子对接结果图的绘制
文献中常见的3D分子对接结果图绘制:利用PyMol和adobe illustrator完成①打开PyMol→“file”→“open”→选择刚才保存的“FHW.pdb”文件→“打开”;②将受体蛋白结构设置为一个整体选项并调整配色:点击操作界面右下角的“SEQ”显示受体蛋白和小分子配体的序列和名称(点击“display”→“sequence mode”→“Residue Names”可以将受体蛋白的序列显示更改为氨基酸残基名称显示),再点击操作界面左上角的“Residues”,选择“Chiains”,然后在显示界面或上方序列处单击选中受体蛋白的整个结构,打开右侧(sele)的“A”选项卡→“rename selection”,这时候在显示界面的左上角会出现一个小窗格“renaming sele to:sele_”,直接点击键盘的“Backspace”删除“sele”输入“protein”将其修改为“renaming sele to:protein_”→“Enter”(这里实际上是将受体蛋白的结构设置为一个整体选项,并命名为protien,方便后续进行快速准确的选择)→打开(protein)的“C”选项卡,根据喜好设置颜色,如spectrum-rainbow(elem c);③将“Chiains”切换回“Residues”→选中小分子配体→打开(sele)的“A”选项卡→“rename selection”,将“renaming sele to:sele_”修改为“renaming sele to:PT_”→“Enter”(同样是将小分子配体的结构设置为一个整体选项,并命名为PT,方便进行快速选择),也可以根据喜好调整配色;④显示小分子配体与受体蛋白之间生成的氢键:打开(PT)的“A”选项卡→“find”→“polar contacts”→“to other atoms in object”,可以看到共有四根氢键生成;⑤分析生成的氢键连接的是受体蛋白上的哪几个氨基酸残基:打开(protein)的“S”选项卡→“sticks”显示受体蛋白结构的棍棒模型以便观察,同时还可以调整受体蛋白和小分子配体结构的显示颜色→在受体蛋白结构中查找并单击选中每一个与氢键相连的氨基酸残基→打开(sele)的“A”选项卡→“rename selection”→“renaming sele to:sele_”修改为“renaming sele to:AJSCJ_”→“Enter”,然后再打开(protein)的“H”选项卡→“sticks”隐藏受体蛋白结构的棍棒模型,接下来再打开(AJSCJ)的“S”选项卡→“sticks”显示氢键连接的两个氨基酸残基的棍棒模型,同时调整这两个氨基酸残基的显示颜色以及背景的颜色(“display”→“background”→“white”)以便观察分析;⑥导出分子对接完整结果图:导出图片时,输出的图片只会包含显示界面中包含的结构部分,所以要使整个蛋白结构位于画面当中,可以点击操作界面上方的“Zoom”,选择“all”,调整至合适角度→点击操作界面右上方的“Draw/Ray”,将分辨率“DPI”的值改为300→“Draw(fast)”→根据需要选择保存为文件(Save image to file)或复制到剪切板(Copy image to clipborad);⑦导出分子对接的对接细节图:通过缩放大小、调整角度和位置等方式,找到一个能够较好显示小分子配体和受体蛋白对接情况的画面,改变受体蛋白结构的透明度以突出显示配体及生成的氢键(点击“setting”→“Transparency”→“Cartoon”→选择合适的透明度),同时还可以调整受体蛋白或配体结构的显示格式(如打开protein的“S”选项卡→“surface”显示表面模型,同时点击“setting”→“Transparency”→“Surface”选择合适的透明度并调整画面大小),当结构模型等调整好之后就可以添加相关的文字和数据批注了,如氨基酸残基名称/氢键长短等(打开AJSCJ的“L”选项卡→“residuce”显示氨基酸残基名称,如果没显示文字可能是被蛋白表面模型覆盖了,直接在最下方的命令框中输入命令“set float_labels,on”即可,然后再打开PT_polar_conts的“S”选项卡→“lables”显示氢键的长度,另外,点击“setting”→“lable”→“Size”可以调整字体大小,点击操作界面上方的“Mouse”→“3 Button Editing”→按住“Ctrl”配合鼠标左键可以拖动文字标签的位置,完成后切换到“3 Button viewing”),再点击操作界面右上方的“Draw/Ray”,将分辨率“DPI”的值改为300→“Draw(fast)”→根据需要选择保存为文件(Save image to file)或复制到剪切板(Copy image to clipborad);⑧完成整体视图和局部视图的拼接:直接在Ai内导入或复制图片,调整大小、设置边框、添加线条等即可。
注:在保存图片时PyMol软件出现了闪退问题,导致对接工作直接中断。首先怀疑可能是分辨率设置过高,所以导入了之前输出的受体-配体复合物结构文件,以150DPI导出仍然闪退。这时候联想到AutoDock vina安装时的闪退问题,怀疑可能是显示适配器被禁用造成的,因此参照禁用步骤启用了显示适配器AMD Radeon(TM) Graphics,再次尝试保存未再出现闪退问题。
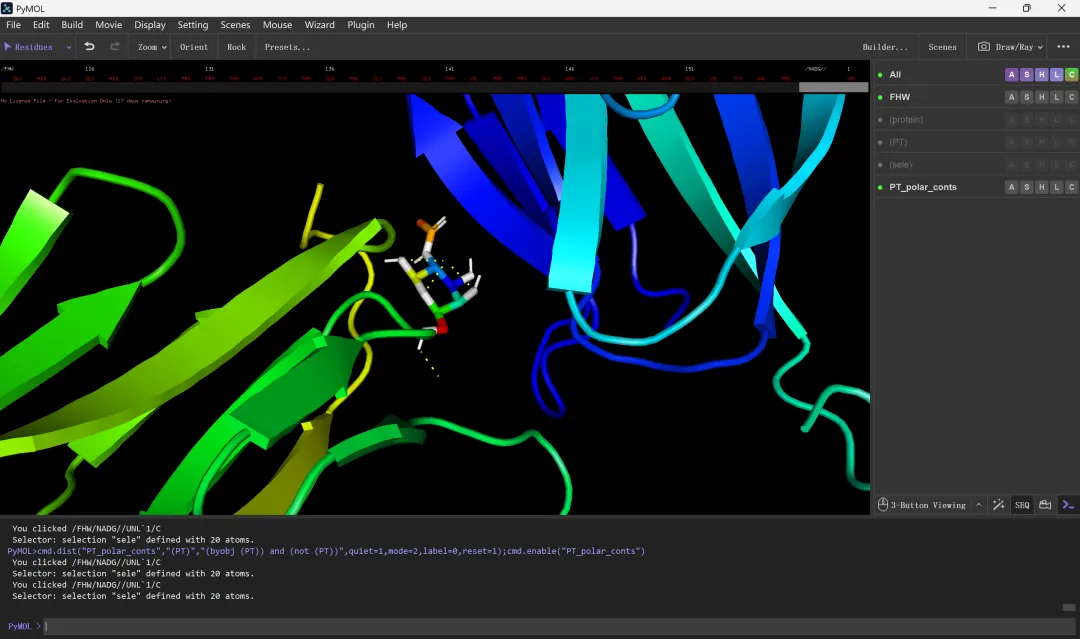
图 19 根据需要调整受体和配体结构的配色
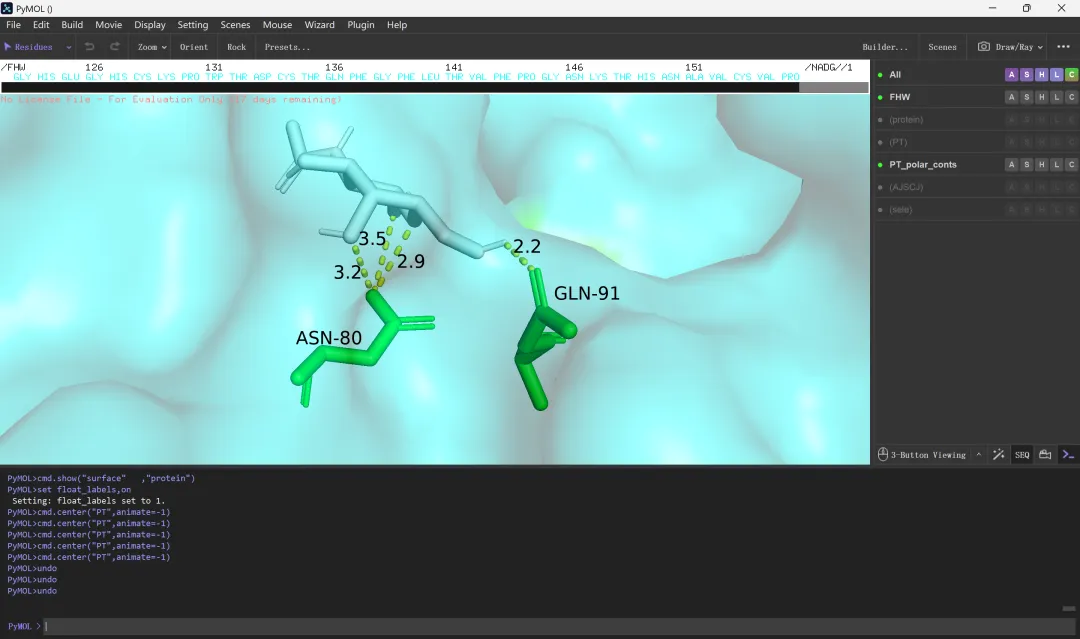
图 20 调整受体和配体的显示并标注残基名称/氢键长度
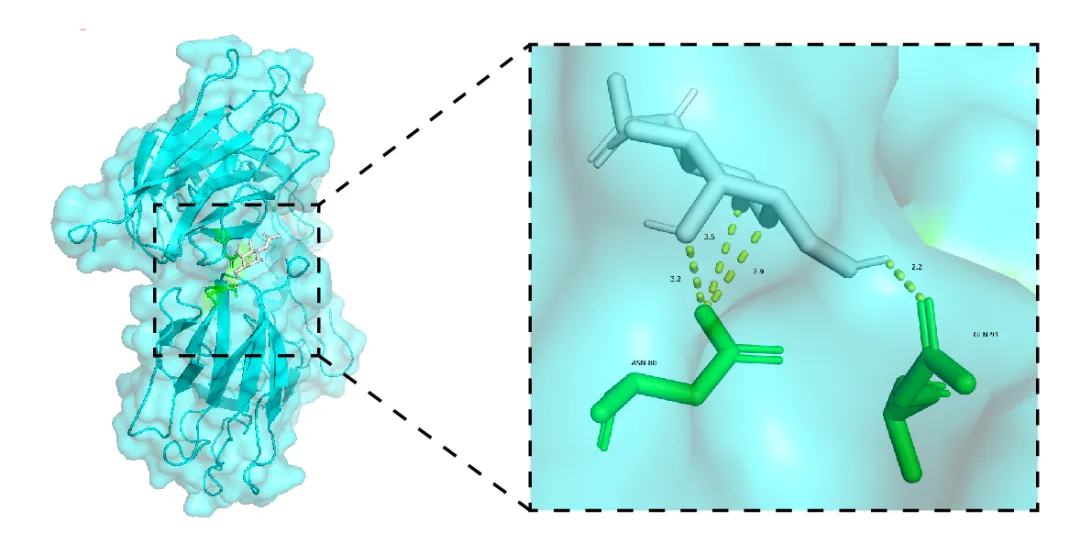
图 21 利用Ai制得简要的分子对接3D结果图(未精调)
文献中常见的2D分子互作图绘制:除了3D分子对接结果图外,文献中一般还会提供2D分子互作图。制作2D分子互作图可以利用LigPlot+完成,具体操作步骤:①首先打开电脑文件资源管理器,在电脑C盘一级目录下创建一个名为“tmp”的空文件夹(临时文件),路径为“C:\tmp”;②打开LigPlot+→“file”→“Open”→“PDB/mmCIF file”→“Browse”→选择“FHW.pdb”分子对接结果文件→“open”→在LIGPLOT栏目下可以选择配体/在DIMPLOT栏目下可以选择需要进行互作分析的蛋白质的两条链/再Antibody栏目下可以设置抗体相关的分析,根据需要进行自由选择,对于受体蛋白和小分子配体之间的互作分析,直接在LIGPLOT栏目下选择好配体,再点击“run”即可加载互作图,之后,点击On/off、Sizes、Colurs等可以设置具体的显示格式和内容,如字体大小、原子大小、填充颜色等,同时图中的文本和结构也可以进行拖动和改动;③导出结果文件:点击“file”→“print screen”可以保存为PDF,“Write PS file”则会保存为PS文件;④在2D分子互作图中,无法观测到π-π共轭等相互作用,因此需要借用PyMol进行三维观测,具体步骤:点击LigPlot+结果界面右下角的“PyMol”(前提是要设置好PyMol的路径)将自动打开PyMol并把2D互作图更改为三维模式,PyMol打开后,可以调节小分子配体的颜色,以便观察。
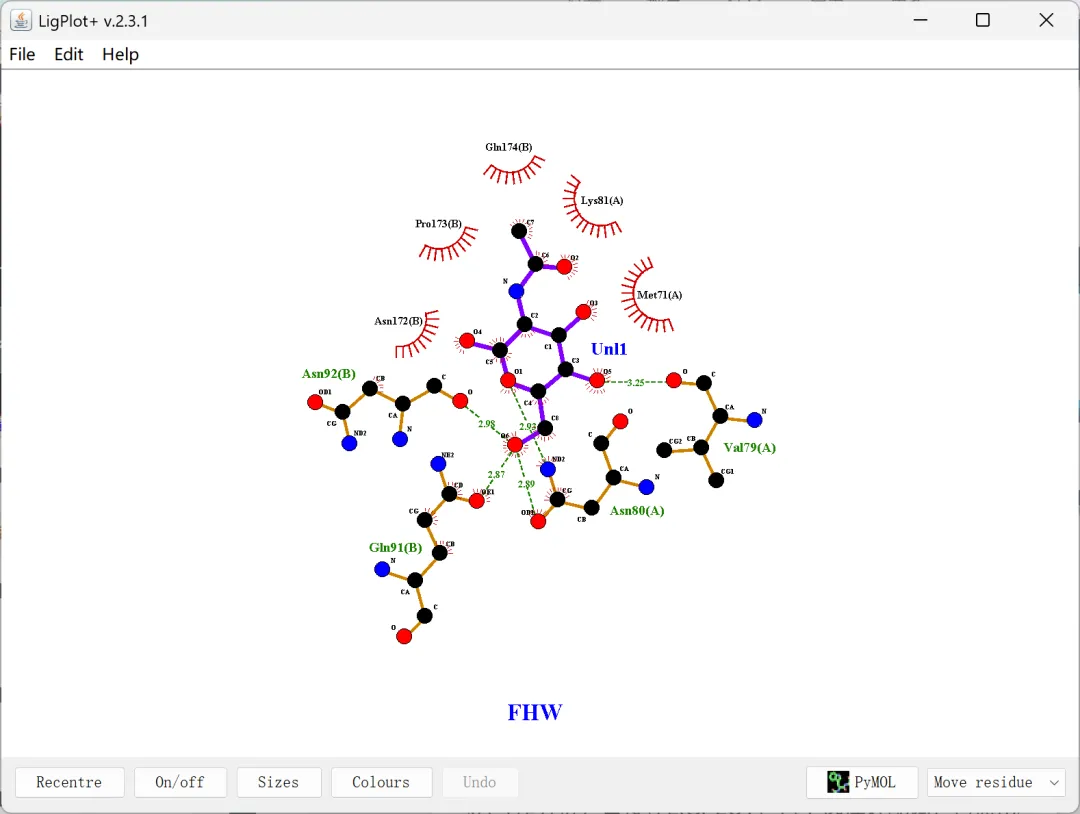
图 22 受体蛋白与小分子配体的2D分子互作图
这里再补充说明一下2D分子互作图中各个元素的具体含义,Ligand bond(配体键)、Non-ligand bond(非配体键)、Hydrogen bond and its length(氢键及其长度)、Non-ligand residues involved in hydrophobic contacts(参与疏水作用的非配体残基)、Corresponding atoms involved in hydrophobic contacts(参与疏水作用的相应原子)。

图 23 2D分子互作图中各元素的含义
(引自LigPlot+官网https://www.ebi.ac.uk/thornton-srv/software/LigPlus/manual2/manual.html)
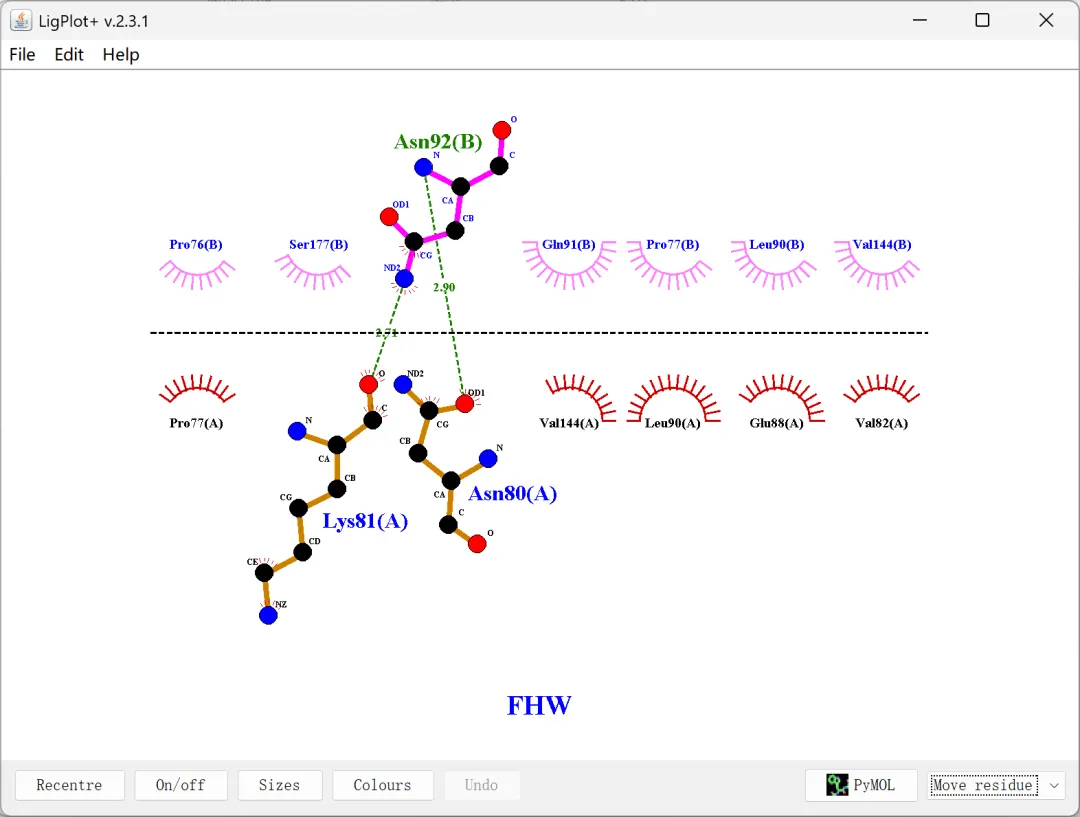
图 24 受体蛋白A链与B链之间的2D互作图
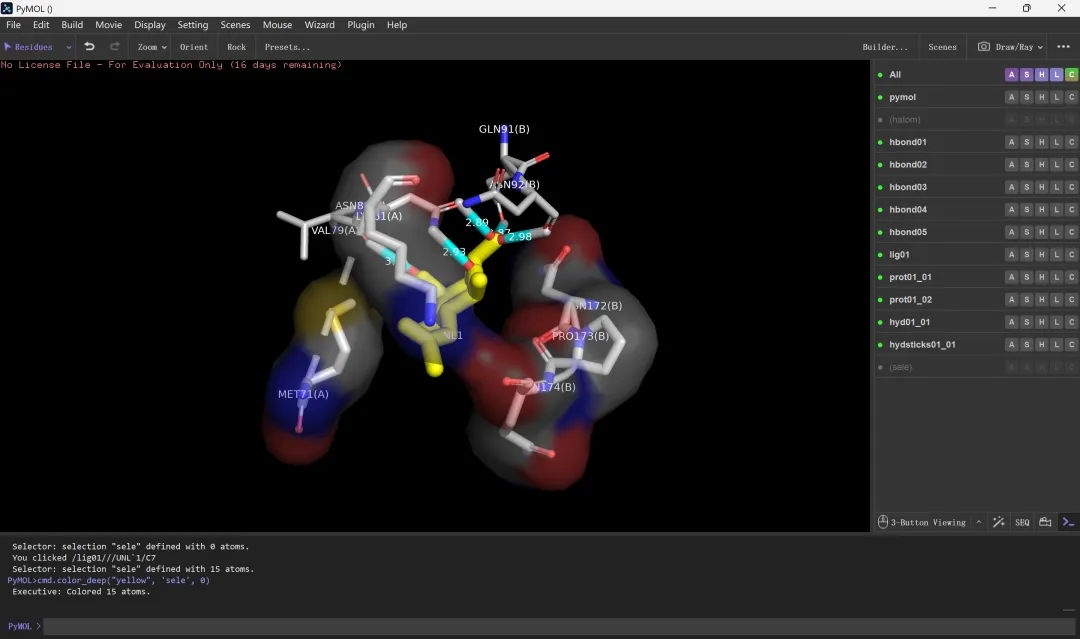
图 25 将2D互作图在PyMol中转换为3D模式(仅供示意)
注:在尝试通过LigPlot+窗口打开PyMol时总是无任何反应(即无法正常链接并调动PyMol),多次重复删除、下载操作及切换安装硬盘均无法解决该问题。询问deepseek得到一种分析和解决方案是“将路径精确指向PyMol的主程序文件(.exe)而不是安装文件”,因此点击“Edit”→“program paths”→将“PyMOL executable”输入框中的路径“D:\PyMol”手动更改为“D:\PyMol\PyMOLWin.exe”,保存后再次尝试打开PyMol,成功链接并调动。
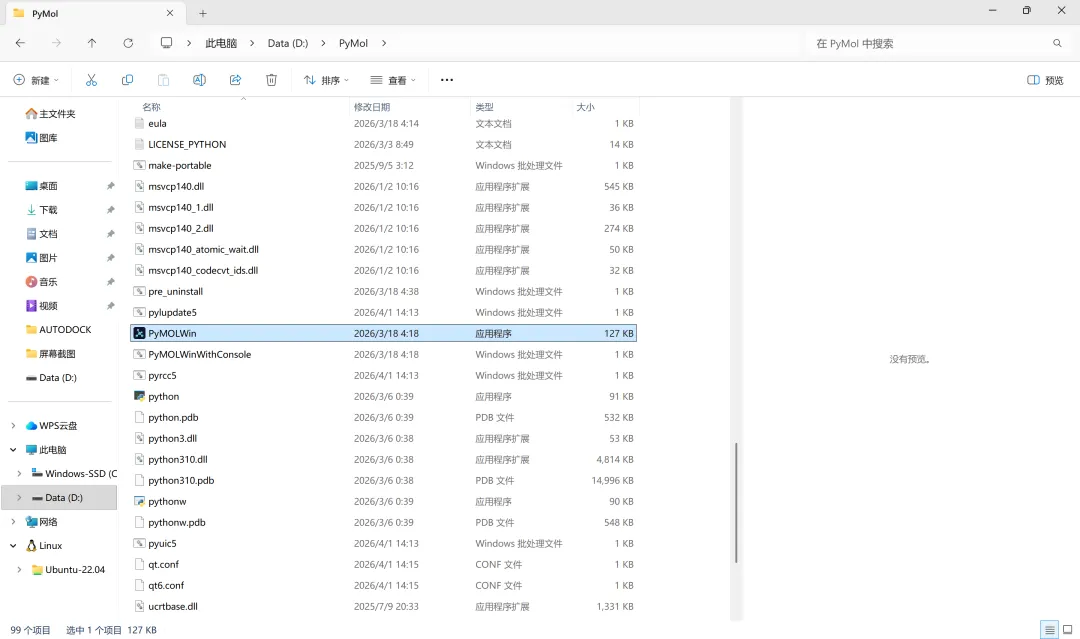
图 26 原本路径指向整个PyMol文件夹而非.exe应用程序
在线分子对接工具
Online molecular docking tools
SwissDock:是由瑞士生物信息学研究所(Swiss Institute of Bioinformatics)开发的免费在线分子对接工具,用于预测蛋白质与小分子的相互作用,支持Attracting Cavities和AutoDock Vina双对接算法。
(1)官方网址:https://www.swissdock.ch/
(2)简要使用步骤:在线分子对接平台的优势在于少了繁琐的软件安装过程,且也有着较为明确的操作指引,因此这里就不再进行详细的介绍了,感兴趣的可以自行了解和学习。
Protein-Ligand Interaction Profiler(PLIP):一款开源且高度自动化的分析工具,能够快速识别蛋白质、DNA 或 RNA 与配体之间的多种非共价相互作用,并提供原子级细节、可视化图像以及标准化输出文件,可用于2D分子互作图的制作。
Mob提示
Mob’s reminder
本期文字内容较多,部分地方语言描述可能不是特别清晰准确,加之展示的大多都是结果图片,在实际操作过程中看起来多少会有一些吃力,但是有任何疑问,随时欢迎在评论区留言或私信咨询。同时,本教程主要介绍分子对接的关键步骤,对于颜色、显示模式等的设置未做过多的展示,根据自己的需要和审美进行灵活调整即可。另外,后期如果时间和精力允许会考虑出视频教程,如有更深入的内容需求也欢迎投稿,感谢支持。
在线学习资源
Online learning resources
私信回复【分子对接】无偿获取本期资料
往期专题学习资料,回复【关键词】无偿获取
(注意字母大小写)
Adobe illustrator制图【Ai制图】
Graph Prism制图【Prism制图】
ChemDraw制图【ChemDraw】
生物信息学与功能基因组学【生信与组学】
R语言生信分析【R语言】
核磁共振氢谱解析【NMR核磁共振】
色谱/质谱原理及识谱【LCMS】
靶向蛋白讲解技术【TPD】
……
长期不定时在线更新
Chem.BM

关注并点赞分享帮我充电,阿里嘎多~