 夜雨聆风
夜雨聆风





进化溯祖 | BEAST相关文档解读
细菌基因组 | BEAST简介及Linux版安装
安装好BEAST的Windows版后,看到以下帮助文档
bin/ 对应操作系统的脚本
文档2 – BEAST的所有文档现已在线 (All documentation for BEAST is now online):http://beast.community/ (发现文档3-BEAST社区)。

文档1
然而,这种灵活性意味着在所使用的马尔可夫链-蒙特卡罗(Markov chain Monte Carlo, MCMC)推理框架下,有可能构建了表现不佳的模型。我们无法测试BEAST中可以使用的所有可能的模型。
对此有两种解决方案:首先,我们为常见的分析提供了一系列方法的组合 (A range of recipes),这些分析可在BEAST中正常工作,并为其提供了示例输入文件 (尽管分析实际-Actual数据时,也可能发生意外)。其次,我们提供了诊断问题的工具,以及解决问题的建议:http://beast.community/
BEAST不是一个”黑匣子”,你可以把你的数据放进去,期待一个容易解释的答案。其需要仔细审查输出,以检查它是否正确执行,通常需要调整、调试并多次运行,才能得到有效结果。
序列转换
一个名为“BEAUti”的程序将以NEXUS格式导入数据 (也能解析FASTA/.aln),来选择各种模型和选项,并生成一个可在BEAST中使用及运行的XML文件。
要运行BEAUti,只需双击“BEAUti v1.10.x”应用程序。若不起作用,那么可能没有正确安装Java。尝试打开MS-DOS窗口并键入命令行:java-jar-lib/beauti.jar。另请参阅单独的:BEAUti – README.txt 文档。
运行BEAST
双击“BEAST v1.10.x”应用程序。将被要求选择一个XML输入文件。
或者打开“命令”窗口并键入:java-jar lib/beast.jar input.xml
其中“input.xml”是BEAST xml格式文件的名称。该文件可使用文本编辑器从头开始创建,也可由BEAUti程序从NEXUS格式文件创建。
有关创建和调整输入文件的文档,请参阅以下联机文档和教程:
帮助 http://beast.bio.ed.ac.uk
常见问题 http://beast.bio.ed.ac.uk/FAQ
教程 http://beast.bio.ed.ac.uk/tutorials
最新手册 code.google.com/p/beast-mcmc
BEAST 参数:
-verbose 输出XML解析信息
-warnings 输出XML警告信息
-strict XML不合格时失败
-window 提供1个控制台窗口
-options 展示1个选项框
-working 切换工作目录至输入文件所在区
–seed 指定随机数生成种子
-prefix 输出文件的前缀
–overwrite 允许覆盖已有文件
-errors 在停止前,指定数值错误的最大数
–threads 线程数 (默认:自动)
-java 只使用Java而非本地实施
-threshold 充分评估测试阈值 (默认:0.1)
–beagle 如果可用,使用BEAGLE库 (默认:on)
-beagle_off 不使用BEAGLE库
-beagle_info BEAGLE: 展示可用资源的信息
-beagle_auto BEAGLE: 自动选择最快的资源来分析
-beagle_order BEAGLE: 设置资源使用的顺序
-beagle_instances BEAGLE: 在实例之间划分位点模式 (Divide site patterns)
-beagle_CPU BEAGLE: 使用CPU实例/Instance
-beagle_GPU BEAGLE: 使用GPU实例,if available
-beagle_SSE BEAGLE: 使用SSE扩展 (与CPU型号有关) (if available)
-beagle_SSE_off BEAGLE: 关闭SSE扩展
-beagle_cuda BEAGLE: 使用CUDA并行,if available
-beagle_opencl BEAGLE: 使用OpenCL并行,if available
-beagle_single BEAGLE: 使用单精度/Single precision,if available
-beagle_double BEAGLE: 使用双精度,if available
-beagle_async BEAGLE: 使用异步内核 (Asynchronous kernels),if available
-beagle_scaling BEAGLE: 指定使用的标化方案 (Scaling scheme)
-beagle_rescale BEAGLE: 重新标化的频率 (只在动态标化下)
–mc3_chains 链 (Chains)的数量
-mc3_delta 温度增量参数
-mc3_temperatures 热链温度(Hot chain temperatures)的逗号分隔列表
-mc3_swap 链温度交换 (Swapped)的频率
-load_dump 指定要从中加载转储状态 (Dumped state)的文件名
-dump_state 指定写入转储文件的状态 (State)
-dump_every 指定写入转储文件的频率 (Frequency)
-version 软件版本
例:
java -jar lib/beast.jar -seed 12345 -overwrite input.xm分析结果
我们制作了一个强大的图形程序来分析MCMC日志文件 (它也可以分析MrBayes和其它MCMC的输出)。其称为“Tracer”,可从Tracer网站获得:tree.bio.ed.ac.uk/software/tracer
我们现在再次纳入了“loganalyser”程序,以便在无需Tracer的情况下,分析日志和树文件。(BEAST v1.10.4/bin/loganalyser.cmd?)
此外,还有两个新程序作为BEAST包的一部分分发:LogCombiner和TreeAnnotator。
LogCombiner可将多次运行BEAST的日志或树文件,合并为一个单独的组合结果文件 (在舍弃适当的”预烧”即Burn-ins后)。
TreeAnnotator可使用单个目标树 (A single target tree),总结BEAST中的树样本 (A sample of trees from BEAST),并用后验概率、HPD结点高度和速率 (Posterior probabilities, HPD node heights and rates)对其进行注释。
然后可以在名为“FigTree”的新程序中查看该树,其可从以下位置获得:tree.bio.ed.ac.uk/software/figtree
原生库 (Native Libraries)
BEAST使用BEAGLE库来提供快速、原生的似然计算。此库是独立分发的,应在使用BEAST之前安装。
支持和链接
BEAST是一个极其复杂的程序,因此不可避免地会出现错误。
请给我们发电子邮件讨论任何问题:alexei@cs.auckland.ac.nz; a.rambaut@ed.ac.uk; msuchard@ucla.edu
文档3

什么是BEAST?
BEAST是一个使用MCMC对分子序列进行贝叶斯分析的跨平台程序。它完全倾向于使用严格或宽松的分子时钟模型,推断的有根的、时间测量的系统发育。
BEAST可以作为一种重建系统发育的方法,但也可以作为一个测试进化假设的框架,而不以单一的树拓扑为条件。BEAST使用MCMC在树空间上进行平均 (Average over tree space),从而使每棵树的加权与其后验概率成比例 (Each tree is weighted proportional to its posterior probability)。
BEAST包含一个简单易用的用户界面程序,用于设置标准分析,以及一套用于分析结果的程序。
此网站 (http://beast.community/)适用于BEAST v1.X (当前版本v1.10.4)。此外,奥克兰大学领导的独立项目BEAST2的详细信息,见:http://beast2.org/。
入门教程
作为使用BEAST的入门,我们提供了一些基本的入门教程,使用BEAST图形应用程序,使用提供的示例文件执行分析。http://beast.community/getting_started
引用BEAST
《使用 BEAST 1.10 进行贝叶斯系统发育和系统发生动力学数据整合》
doi: 10.1093/ve/vey016
BEAST源自早期的大量工作。BEAST建立在大量先前工作的基础上,运行BEAST时,将列出对单个模块、模型和组件的适当引用:
Pybus OG & Rambaut A (2002) GENIE: estimating demographic history from molecular phylogenies. GENIE:从分子系统发育中估计种群历史。Bioinformatics, 18, 1404-1405.
文档4
beast.community/getting_started
下载并安装BEAST。需安装两个额外的软件包,是BEAST分析的关键部分。即:Tracer,用于分析BEAST的输出;以及FigTree,用于查看生成的树,并为发表做好准备。
教程1。介绍了使用BEAUti和BEAST来分析一些灵长类动物序列并估计系统发育树。它将引导您完成导入数据、选择模型、生成 BEAST XML 文件的过程。然后运行BEAST。
教程2。本教程直接从第一个开始,查看BEAST的输出,诊断问题并总结结果。本教程将介绍一些与BEAST、LogCombiner和TreeAnnotator打包的实用程序,以及外部应用程序Tracer和FigTree。
估算 (进化)速率和日期。本教程提供了一个循序渐进的教程,用于分析在不同时间点分离的一组病毒序列 (即异时数据)。这项工作的目的是获得分子进化率的估计,最近共同祖先的日期的估计,并通过适当的统计支持措施,推断系统发育关系。
可在这里找到教程和具体“操作”说明的完整列表:
http://beast.community/tutorial_contents
文档5
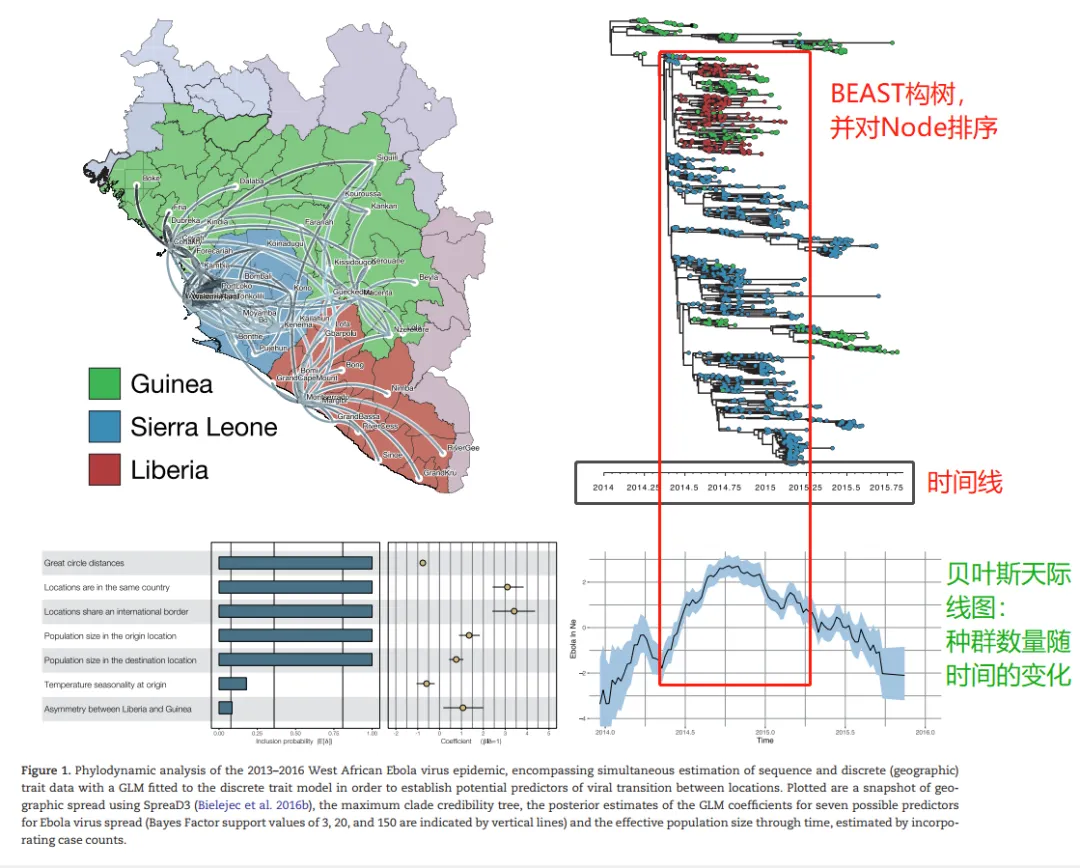
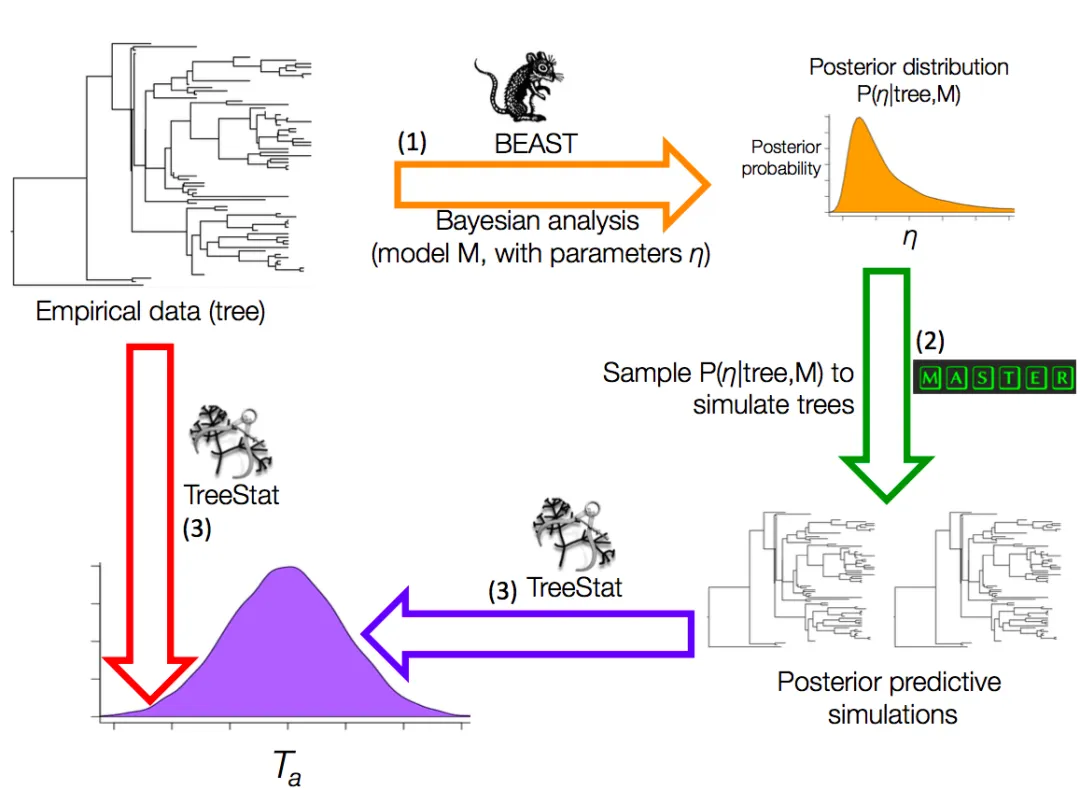
www.beast2.org/2018/07/24/TreeModelAdequacy.html
扫码、联系客服老师报名,领取资料、上手分析
