 夜雨聆风
夜雨聆风





筛多肽的三把刀:AI预测、分子对接、QSAR,到底该怎么用?
同样是筛多肽,为什么有人三个月出一条候选、有人三年还在原地打转?区别往往不在工具本身,而在有没有用对工具,以及有没有把工具串成一条线。
一个真实的困惑
最近和几位做功能性多肽(抗炎、抗衰、抗菌方向)的朋友聊天,发现一个普遍现象:
大家都知道现在有三类主流方法可以做多肽筛选——机器学习预测、分子对接、QSAR。但真正动手时,问题就来了:
-
是先做ML预测,还是先做对接?
-
我手上没有靶点,能不能直接做对接?
-
QSAR听起来很老派,现在还有用吗?
-
三个方法是分着用,还是组合着用?
-
AlphaFold3、RFdiffusion这些新东西出来以后,老流程还要不要走?
更尴尬的是,很多人花了大量时间在一个方法上反复打磨,最后发现选错了路径——手上的数据和问题,根本不该用那把刀。
这篇文章就来把这件事讲清楚。
先打个比方:三把刀分别是干什么的
为了让后面的讨论更直观,我们先做一个类比。假设你是一家猎头公司老板,要从100万份简历里找到最适合某个岗位的人选:
方法一·机器学习预测,相当于你雇了一个经验丰富的HR。她不需要见到求职者本人,只看简历上的关键词、学历、经历,就能凭经验给每份简历打分。优点是快——一天看10万份不在话下;缺点是她的判断完全基于”过去成功的人长什么样”。如今HR还进化出了一种”AI画像师”模式(生成式AI)——不再是被动看简历,而是直接根据岗位需求画出理想候选人的样貌,让你照着去找。
方法二·分子对接(含复合物结构预测),相当于你安排了一场线下面试。求职者本人到场,和岗位的实际场景做模拟匹配,看他们站在一起合不合拍。优点是直观、有解释力——你能看到”为什么这个人合适”;缺点是慢,而且前提是你得先有一个明确的岗位。
方法三·QSAR(含其现代变体),相当于你已经有了一个还不错的候选人,现在想做”职业咨询”——告诉他”你把这段经历换个写法、再考个证书,offer机会能涨50%”。它解决的不是”找人”的问题,而是”把已有的人优化得更好“。
理解了这个类比,我们再看这三把刀在多肽筛选里到底各自能做什么、不能做什么。
第一把刀:机器学习预测——广撒网,秒级筛选
它在解决什么问题
ML方法的核心逻辑非常朴素:收集大量已知活性的多肽数据,让模型学会”什么样的序列容易有活性”,然后拿去预测新序列。
具体来说,你会从公共数据库(比如抗菌肽的APD3、DRAMP,抗炎肽的IEDB-AIE子集,AntiInflam等预测工具的训练集,抗衰肽的BIOPEP-UWM等)拉来几千上万条带活性标签的多肽,把序列丢进ESM-2、ProtBERT这类预训练蛋白语言模型做向量化,然后接一个XGBoost或者轻量级神经网络做分类(是/不是抗炎肽)或回归(活性多强)。
它的甜点场景
当你手上有很大的候选库、但暂时不需要管机制时,ML是唯一现实的选择。
举一个2021年IBM团队发在 Nature Biomedical Engineering 上的经典案例:他们用深度生成模型(CLaSS方法)生成了大约9万条候选肽序列,再用深度学习分类器+粗粒度MD做多级筛选,最终合成测试20条,其中2条对包括耐药肺炎克雷伯菌在内的多种Gram阳性/阴性菌都有效,整个流程48天走完。
注意——这里很多科普文流传的”8000万条候选”是讹传,原始论文和C&EN等媒体报道的实际数字是9万级别。真正达到亿级筛选规模的工作另有其例(如最近用 PeptideAtlas 5.5M 库的 LLAMP,以及 ProteoGPT 系列),但都是更晚的工作。
ML的新形态:从”筛”到”造”
只把ML当作分类器/回归器,是2020年前的思路。2026年的主流玩家已经在用生成式AI直接画候选肽——
-
序列空间生成:ProtGPT2、AMP-Designer 这类基于蛋白语言模型的生成器,可以根据指定属性(如抗菌+低毒+特定靶点)直接产出全新序列;
-
结构空间生成:RFdiffusion、ProteinMPNN 这类扩散/反向折叠模型,可以先指定结合口袋,再倒推出能填进去的多肽骨架——这意味着你甚至不需要先有真实的活性数据。
生成式方法部分绕开了”训练集外推差”的老问题,因为它本质上是在物理/几何约束下采样,而不是在历史数据里插值。
它的天花板
但ML(包括生成式)仍有几个绕不开的问题:
第一,可解释性弱。模型告诉你”这条肽是抗炎肽,置信度0.92″,但说不清为什么。SHAP、Attention可视化、AlphaFold结构等可以缓解一部分,但远不如直接的结合姿态图直观。
第二,过度依赖数据质量。Garbage in, garbage out——抗炎肽数据库里大量是”活性级别从μM到mM都混在一起”的脏标签,模型学完后给的”活性预测”经常没有意义。自建数据库的标注一致性,决定了模型的天花板。
第三,容易筛出”对的但不能用的”——比如抗菌活性很强但溶血性也爆表的肽,或者活性预测很高但分子量太大、根本透不过靶细胞膜的肽。这部分要靠后面的多目标过滤(毒性预测、理化性质预测)补救。
一句话总结
ML是漏斗的最上层——负责把100万缩到1000,必要时还能”凭空生成”候选;但需要靠下游工具回答”为什么好”和”能不能用”。
第二把刀:分子对接与复合物结构预测——看清”怎么结合”
它在解决什么问题
对接的逻辑和ML完全相反:它不需要任何活性数据,但必须有靶点的三维结构。
传统流程是:你给它一个多肽(用AlphaFold3、ESMFold等预测出3D结构)和一个靶蛋白(比如TNF-α、IL-6R、SIRT1、MMP-9),对接软件(HADDOCK、Rosetta FlexPepDock、AutoDock CrankPep等)会尝试上千种结合姿态,找出能量最低的那个,然后输出结合自由能ΔG作为亲和力打分。
一个重要的现状变化:AF3 正在吞掉传统对接
到2026年,对短肽-蛋白复合物,AlphaFold3(以及Boltz-1、Chai-1等同代模型)的精度已经显著超过经典对接流程。原因有两个:
-
AF3 直接联合预测复合物结构,避免了”先单独预测多肽构象、再硬塞进口袋”的两步累计误差;
-
它内部学到了大量蛋白-蛋白接触模式,对PPI界面的预测尤其稳定。
所以现在的合理做法是:先用 AF3 做复合物结构预测,再把得到的结合姿态送到 Rosetta/HADDOCK 做局部优化和重打分。纯传统对接软件的角色越来越像”细化工具”,而不是主筛工具。
它的甜点场景
靶点明确、机制清楚、候选肽不多的时候,结构预测+对接是最优解。
抗炎方向是最典型的例子:TNF-α是一个三聚体,它和受体的结合界面是一个相对平坦的PPI界面。这种界面的特点是——纯ML几乎学不会,因为它涉及精细的空间互补。这时候用 AF3 / AlphaFold-Multimer 预测多肽-TNF-α复合物,再用 MM/PBSA 重打分,是远比纯ML靠谱的策略,相关文献近几年很多。
更重要的是,结构预测的产出是可视化的结合姿态图——你能清楚看到”这条肽的第3位精氨酸插入了TNF-α的酸性口袋”。这张图既能解释机制,又能直接拿去申专利、画毕业论文。
它的天花板
但对接/结构预测也有它的坑:
第一,长柔性肽仍然难。短的、刚性的、有明显二级结构的肽(α螺旋、β发夹)效果最好。一旦肽超过20个氨基酸、且本身在水中是无序的,AF3的pLDDT/PAE置信度会明显下降。
第二,打分函数 ≠ 实际活性。无论是经典对接的ΔG、还是AF3的ipTM/PAE,它们和实测IC50/KD的相关性通常只在0.3-0.6之间。所以业内有句话:”对接看的是排名,不是绝对值。”想拿到更可靠的能量排序,还得靠 MM/PBSA、FEP+ 这类后处理。
第三,算力不便宜。AF3 跑一个复合物,单卡A100大概几分钟到几十分钟。几千条肽全跑一遍,账单很快上去。
一句话总结
对接(含AF3级结构预测)是漏斗的中层——负责把1000缩到50,并且告诉你”为什么这50条好”。
第三把刀:QSAR——已经知道方向后,告诉你怎么改
它在解决什么问题
QSAR(定量构效关系)的核心逻辑是:
在一组结构相似的多肽里(比如同一个母核的衍生物),把分子的描述符(电荷、疏水性、3D电场等)和活性做定量回归,找出”哪些结构特征决定活性”。
经典QSAR包括CoMFA、CoMSIA、HQSAR等。当它和对接结合时,可以在已知的结合构象周围做3D-QSAR——相当于在结合口袋里画一张”等势线图”,告诉你”这个位置加正电荷活性会涨、那个位置加大体积基团活性会降”。
它的甜点场景
当你已经有了一条先导多肽(lead),想做精细优化时,QSAR是黄金工具。
最经典的案例之一是ACE抑制肽(降血压方向)。1990年代,日本Calpis等团队从酪蛋白(不是乳清)发酵物里直接分离到了 IPP(Ile-Pro-Pro)和 VPP(Val-Pro-Pro),随后这两个肽被商品化用于功能性食品(如 Calpis 的 Ameal S 系列酸奶饮料)。
真正展示QSAR价值的不是这两个先导肽本身,而是它们之后的工作:研究者基于IPP/VPP做QSAR分析,发现了几条规律——
-
C端的疏水性残基(Trp/Pro/Phe)是关键决定因素;
-
N端正电荷有利于结合;
-
中间位置对体积比较敏感。
基于这些规律,后来又设计出了 LRW、IKP、FW 等更强的肽(活性比VPP/IPP高一个数量级以上)。QSAR的价值在”先导肽之后的精修”,而不是”凭空设计先导肽”。
抗衰领域的一个常见误读
抗衰领域大名鼎鼎的 Argireline(乙酰基六肽-3,Ac-EEMQRR-NH₂) 经常被科普文当作QSAR案例引用。其实2002年Blanes-Mira等人的原始论文明确把它定性为 “rational design programme”(理性设计)的产物 ——基于SNAP-25 N端序列的功能模拟,而不是经典QSAR回归。N端乙酰化主要是为了提升肽的稳定性和经皮渗透,并不是QSAR告诉化学家”这里加乙酰基活性更高”。
把Argireline列入”基于机制的肽模拟(peptide mimetics)”或者”理性设计”案例更准确。它和QSAR属于不同思路。
QSAR 的现代变体
经典 3D-QSAR(CoMFA/CoMSIA)实操起来很痛苦——分子叠合(alignment)那一步往往把人折磨疯。所以现在的”高级玩家”更多在用:
-
GNN-based SAR:把肽建模成图,用图神经网络直接学结构-活性关系,不需要手动叠合;
-
贝叶斯优化(Bayesian Optimization):少量实验数据迭代地告诉你”下一批合成什么”,是Active Learning流程的核心;
-
Free-Wilson 类加和模型:在同系列衍生物上仍然好用,且解释性强。
如果你看到一篇QSAR论文还在用CoMFA且没有补充任何现代方法对照,多半已经过时了。
它的天花板
QSAR(含现代变体)的局限性:
第一,主要在同系列内有效。如果你的多肽结构差异很大(线性肽、环肽、D型氨基酸修饰肽混在一起),传统QSAR模型直接失效;GNN稍好但也有限。
第二,数据要求。传统3D-QSAR的甜点是几十到几百条同系列衍生物;现代深度学习SAR可以吃下几千上万条,但异质性必须可控。
第三,它本质上是优化器,不是发现器。它擅长”在已有方向上精修”,不擅长”找新方向”。
一句话总结
QSAR(含现代变体)是漏斗的最底层——负责把”还不错的50条”打磨成”非常好的5条”。
容易被忽视的第四把刀:分子动力学(MD)
原版只把MD当作”最后验证一下”,其实低估了它。多肽和小分子最大的不同是——多肽在水里通常是无序的(random coil),只有结合在靶点上才形成稳定构象。这件事单靠静态对接根本看不出来。
MD能补三件事:
-
结合的动力学持久性:解离速率(k_off)往往比瞬间结合力更决定真实活性,尤其是抗炎/抗衰这种长效场景;
-
构象选择:肽在结合前/后构象差异多大、构象熵代价多少;
-
隐藏口袋:μs级模拟有时能揭示AF3静态结构看不到的瞬时口袋。
所以MD应该和对接一起放在”漏斗中层”,而不是只在最后做个收官。
三把刀(+MD)的本质区别:一张表看懂
|
维度 |
ML预测 / 生成式AI |
对接 + 结构预测 |
QSAR / 现代SAR |
MD |
|---|---|---|---|---|
|
核心逻辑 |
序列→活性的统计映射;或物理约束下的生成 |
结构→物理打分 |
描述符→活性的定量回归 |
时序物理模拟 |
|
是否需要靶点 |
不需要(生成式可加靶点约束) |
必须有 |
隐含需要(同系列默认同靶点) |
需要(基于复合物) |
|
数据量要求 |
大(千~万条) |
单次计算即可 |
中(几十~几千条同系列) |
单次计算即可 |
|
可解释性 |
弱~中(视方法) |
强(可视化口袋) |
强(描述符权重明确) |
强(可看动力学) |
|
适合阶段 |
早期广筛/生成 |
中期精筛+机制 |
后期lead优化 |
中后期验证+排序 |
|
典型工具 |
ESM-2、AMPlify、RFdiffusion、ProteinMPNN |
AlphaFold3、HADDOCK、Rosetta |
CoMFA、GNN-SAR、Bayesian Opt |
GROMACS、AMBER、OpenMM |
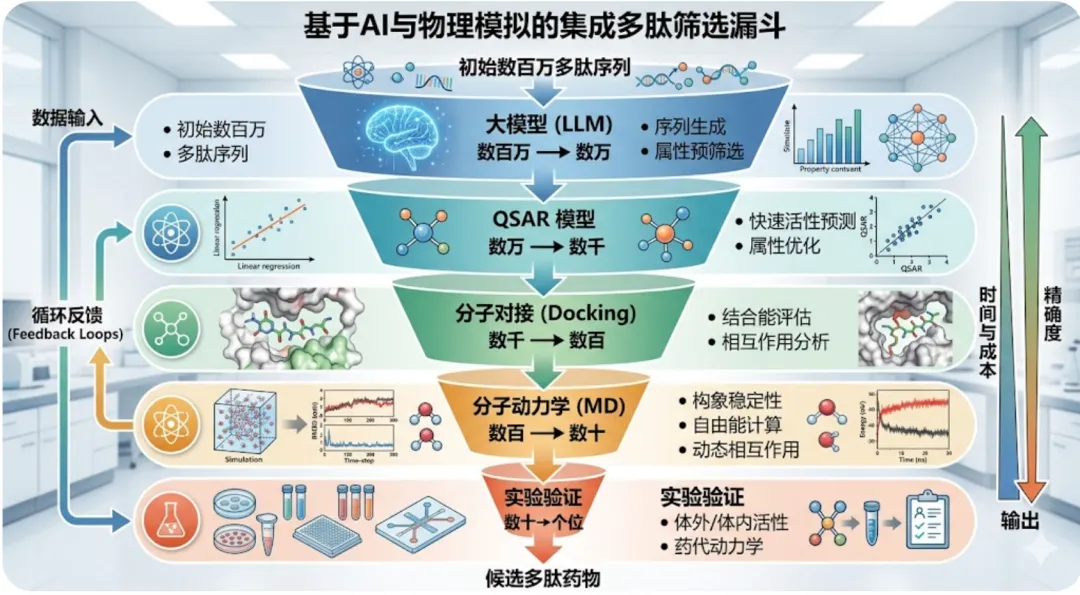
真正高手的玩法:三者(+MD)串成一条漏斗
讲到这里,你应该已经感觉到了——这些方法不是互斥的,而是天然适合串联。
成熟的多肽研发管线一般长这样:
第一关·ML广筛 / 生成:从公共库 + 自建组合库出发,凑出几十万到上百万条候选;或者直接用RFdiffusion/ProteinMPNN生成。用ML模型秒级打分,**同时过掉毒性预测、溶血性预测、理化性质(ClogP、等电点、渗透性)**这些”一票否决”项。剩下几千条。
第二关·结构预测精筛:把幸存的几千条用 AF3 / AlphaFold-Multimer 做复合物结构预测,按 ipTM / PAE / 结合自由能排序,再用 MM/PBSA 重打分,筛出 top 50-100。同时拿到结合模式可视化,机制图就有了。
第三关·MD 动力学验证:对top候选做100ns到μs级模拟,看复合物是否稳定、k_off大概什么量级。这一步会刷掉一些”对接看着好但根本结合不住”的假阳性。
第四关·QSAR / SAR 精修:对剩下的候选做3D-QSAR或GNN-SAR分析,画出等势线图或重要性热图,直接指导化学合成——”第5位Lys换成Arg、第8位Phe换成Trp、N端加乙酰基”。
第五关·闭环反馈(Active Learning):湿实验拿到的真实活性数据,回过头去重新训练第一关的ML模型;贝叶斯优化器告诉你下一批合成哪些。模型越来越准,每一轮筛选效率越来越高。
ML解决”广度”问题,对接/AF3解决”深度”问题,MD解决”持久性”问题,QSAR解决”精度”问题。
而真正能让管线跑起来的是第五关的湿实验闭环——没有真实数据回流,前面四关都是空中楼阁。
不同方向的实战策略
最后给大家一个按方向分的速查表:
抗菌肽方向
抗菌肽的作用机制不是只有膜破坏——APD3现在已经按机制分为攻击细胞壁、碳水、核酸、核糖体等多类。但确实大量天然AMP的主要机制是膜扰动:
-
ML/生成式AI权重最大(IBM案例就是典型)
-
对接价值取决于具体机制:膜作用类用粗粒度MD更合适,胞内靶点(如LPS、脂质II、特定胞内酶)可以做对接
-
QSAR在物化描述符层面(净电荷、两亲性、α螺旋倾向)很有用
-
必做:溶血性预测 + 真核细胞毒性预测。AMP最怕”杀敌一千自损八百”,这两个模型不上,筛出来的全是毒药
抗炎肽方向
抗炎靶点很清晰(TNF-α、IL-6R、NF-κB通路相关蛋白),且大多是PPI界面:
-
AF3复合物预测权重最大——传统对接已经在退场
-
ML做前期粗筛(IEDB AIE子集是常用训练集)
-
QSAR/MD用于先导肽优化和持久性评估
-
注意:抗炎肽的口服稳定性、血脑屏障穿透性(如果是中枢炎症)是后期必须考虑的属性
抗衰肽方向
抗衰是最复杂的,因为”衰老”是多机制的(胶原降解、氧化应激、SASP炎症、神经肌肉传递、端粒维持等):
-
多靶点并行:胶原合成走TGF-β/MMP对接、抗氧化走自由基清除ML回归、类肉毒素效应走SNAP-25模拟
-
通常一个产品需要多条肽组合,每条肽对应一个机制
-
皮肤渗透性是化妆品级肽的最大瓶颈——你筛出的肽活性再强,透不过角质层就是白搭。Argireline的实际功效争议大半来自这里。在管线里必须加入 logP、分子量、TPSA、渗透性预测,且真实皮肤模型(如重建皮肤)的湿实验不可省
写在最后:选对刀,比磨刀更重要
回到开头那个问题——为什么有人三个月出候选,有人三年还在原地?
答案其实很简单:他们手上的问题、数据、目标,从一开始就决定了哪把刀该先用、哪把刀该后用。
-
手上有大数据但没靶点 → 先ML / 生成式AI
-
靶点明确但候选肽少 → 直接 AF3 + 对接
-
已有先导肽要优化 → QSAR / GNN-SAR + MD
-
做完整研发管线 → 四把刀按顺序全用 + 湿实验闭环
工具本身没有高低之分,用对场景才是关键。同样重要的是几条容易被忽视的常识——
-
数据质量决定模型上限:自建库的标注一致性、阴阳性平衡,比换更fancy的模型重要得多
-
多目标过滤比单目标活性预测更接近真实:活性、毒性、稳定性、可成药性必须一起考虑
-
没有湿实验闭环的AI管线都是PPT:哪怕预测的IC50再准,没有回流数据,就没有迭代
希望这篇文章能帮你在下一次开组会、写标书、做项目规划时,少走一些弯路。
如果你正在做某个具体方向(抗炎/抗衰/抗菌)的多肽筛选,欢迎留言交流具体的数据库选择、工具组合和起步管线——我们后续可以针对性地写更细的实操文章。