 夜雨聆风
夜雨聆风





历时两年多,139页PDF回复审稿质疑!大连化物所葛庆杰/孙剑最新Nature 正刊丨羟基诱导钴锰氧化物,温和条件下合成气高选择性制低碳烯烃

题目:Hydroxy-induced cobalt oxides for syngas to light olefins
第一作者: Yu Han (韩誉)通讯作者: Qingjie Ge(葛庆杰),Jian Sun(孙剑)通讯单位:中国科学院大连化学物理研究所论文DOI: 10.1038/s41586-026-10204-4
原文链接:https://www.nature.com/articles/s41586-026-10204-4
研究背景
低碳烯烃(乙烯、丙烯、丁烯)是现代化工的核心基石,广泛用于生产塑料、纤维、橡胶、溶剂及多种精细化学品,全球年需求量超过3亿吨。长期以来,烯烃生产高度依赖石油路线——石脑油或轻烃的蒸汽裂解。该过程需要在800 °C以上的高温下进行,能耗极高,且每生产1吨烯烃约排放1.5–2吨CO₂。随着石油资源日益紧缺和全球“双碳”目标的推进,开发以煤、天然气、生物质等非石油资源为原料经由合成气(CO+H₂)直接制取低碳烯烃的替代路线,成为C1化学与催化领域长达数十年的重大课题。
合成气制烯烃主要有两条技术路线:
- 双功能金属氧化物–分子筛(OXZEO)路线:金属氧化物(如ZnCrOx、MnGaOx等)活化CO和H₂生成酮烯或甲醇中间体,中间体在分子筛(如SAPO-34、AlPO-18)的限域孔道中发生C–C偶联生成烯烃。该路线可实现高达80%以上的低碳烯烃选择性,但通常需要高温(>410 °C)和高压(~6 MPa),且H₂/CO比偏高(~2.5),导致能耗、设备投资和碳排放居高不下,制约了其经济性。
- 费托合成制烯烃(FTO)路线:采用Fe基或Co基催化剂,在相对温和的条件(250–350 °C,0.1–2 MPa)下运行。2016年,包信和、潘秀莲团队发现棱柱状Co₂C纳米晶(暴露特定晶面)可在250 °C、常压下实现61%的低碳烯烃选择性,但CO转化率仅约32%(Nature, 2016, 538, 84)。2022年,该团队通过物理混合疏水聚合物聚二乙烯基苯(PDVB),促进了反应生成的水从催化剂表面快速脱附,将CO转化率提升至64%、烯烃选择性71%(Science, 2022, 377, 406)。疏水策略主要影响水汽平衡,但并未从根本上改变Co₂C表面的CO活化与C–C偶联之间的内在竞争关系。
核心瓶颈:在Co基FTO催化剂中,CO活化与C–C偶联共用同一活性表面(通常是Co₂C)。一方面,CO需要解离生成CHₓ单体;另一方面,这些单体需要发生C–C偶联并控制加氢深度以保留烯键。两者之间存在典型的活性–选择性博弈:过度强化CO活化会导致甲烷和石蜡生成增加,降低烯烃选择性;而抑制CO活化又会导致转化率不足。因此,如何通过新的策略解耦或优化这两个过程,是打破该瓶颈的关键。
大连化学物理研究所葛庆杰/孙剑等的研究提出了一条全新的思路:引入羟基促进剂,诱导生成三斜晶系钴锰氧化物(anorthic Co-Mn oxides)作为CO活化的主要活性相,并与Co₂C协同,使CO活化从直接解离路径转向吸附氢辅助路径。该路径生成含氧中间体(如CHₓO),有利于后续C–C偶联生成烯烃而非过度加氢。这一“亲水促进”策略与疏水策略形成鲜明互补,为温和条件下合成气制烯烃开辟了新方向。
全文速览
本研究报道了一种羟基促进策略,通过将羟基磷灰石(Ca₅(PO₄)₃(OH),HAP)、气相二氧化硅(SiO₂(F))或勃姆石(AlO(OH),AB)等羟基供体与Na₂CO₃修饰的Co₂MnO₄(NCM)前驱体物理混合,在合成气转化过程中原位诱导形成三斜相钴锰氧化物和Co₂C,二者协同催化。在250–260 °C、0.1 MPa、H₂/CO=1–2的温和条件下,该催化剂实现了70–82%的CO转化率和超过60%的低碳烯烃选择性,低碳烯烃碳利用率高达13%,处于同类报道的领先水平。
结合中子衍射、原位XRD/XAFS/XPS、原位TEM、原位DRIFTS和DFT计算,研究揭示:羟基促进剂抑制了Co-Mn氧化物的过度还原(避免生成Co⁰)和深度碳化,稳定了低对称性三斜晶系氧化物相;该氧化物相通过吸附氢辅助的CO解离路径(能垒1.243 eV)生成CHₓO中间体,然后在Co₂C或Co₂C-氧化物界面发生C–C偶联生成烯烃。这一机制从根本上解决了传统FTO中直接解离路径(能垒>1.7 eV)带来的过度加氢问题。该策略还成功拓展至不同羟基供体、不同碱金属助剂和不同Co/Mn比例,展示了良好的普适性。
值得注意的是,从Peer review文件可以看到,该研究工作在投稿阶段经历了多轮严谨的学术评议。例如,审稿人2不认同“羟基”促进作用是提高一氧化碳转化率的直接原因,并且认为它可用于将一氧化碳活化和碳–碳键形成过程分别在不同的位置进行分离。此外,审稿人不明白“羟基”促进剂是如何能够被整合到钴氧化物/结构的主体部分或表面的,进而促进吸附氢辅助的一氧化碳解离的。另外,审稿人也不同意作者关于其研究结果对在温和条件下转化合成气的潜在影响的观点。
为回应质疑,作者以139 页 PDF 呈现了大量补充实验与数据分析,对争议点逐一解答。文章在经过多轮审稿后,最终被顺利接收并发表。例如,作者通过原位 DRIFTS(图 4a – b)、CO 吸附 DRIFTS(补充图 43)以及 DFT 计算结果(图 4c、补充图 45 – 46)清晰地展示了 NCM 和 NCM-OP15 催化剂上 CO 吸附和活化机制所涉及的不同中间体。此外,动力学结果为羟基诱导的 CoxMn1-xO(斜方晶型)与 Co2C(图 R14 – R15)之间的协同效应的有效性提供了更多证据。还添加了一个示意图(图 R13)以进一步阐明我们的核心创新点。
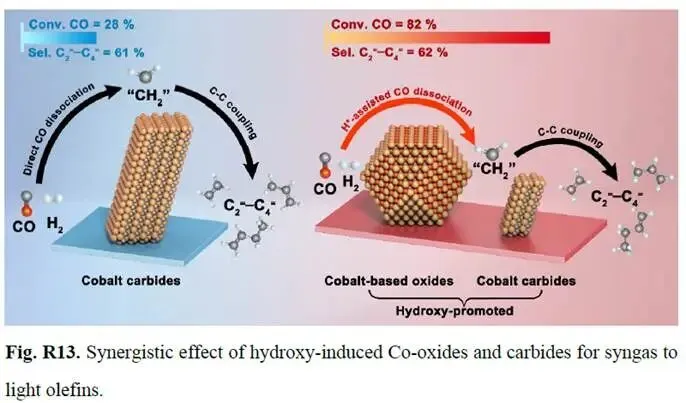
文章亮点
- 首创“羟基促进”新策略:首次发现羟基磷灰石、气相二氧化硅、勃姆石等羟基供体与Co₂MnO₄物理混合后,可诱导三斜晶系Co-Mn氧化物活性相,实现温和条件下合成气高效转化,与疏水策略形成原理性互补。
- 突破活性–选择性博弈:在250 °C、常压、H₂/CO=1–2条件下,CO转化率高达82%,低碳烯烃选择性>60%,碳利用率13%,为同类报道最高之一。
- 揭示三斜晶系相Co-Mn氧化物为CO活化活性相:通过中子衍射、XRD、XAFS、XPS等多技术联用,发现羟基促进剂抑制了氧化物还原为Co⁰和碳化为Co₂C,稳定了低对称性三斜相。该相通过H*辅助CO解离路径活化CO,能垒显著低于直接解离。
- 原位可视化结构演变:在H₂气氛下的原位TEM直接观察到Co₃MnO₄向三斜相Co-Mn氧化物的拓扑转变,同时伴随相邻SiO₂(F)层的薄化和迁移,揭示了羟基促进剂的界面锚定机制。
- 机理清晰、普适性强:DFT计算与实验一致表明,三斜相Co-Mn氧化物表面优先发生H*辅助CO解离,生成含氧中间体,有利于烯烃生成。该策略可拓展至不同羟基供体、不同碱金属和不同Co/Mn比。
图文解析
图1. 羟基促进剂诱导NCM催化剂的催化性能
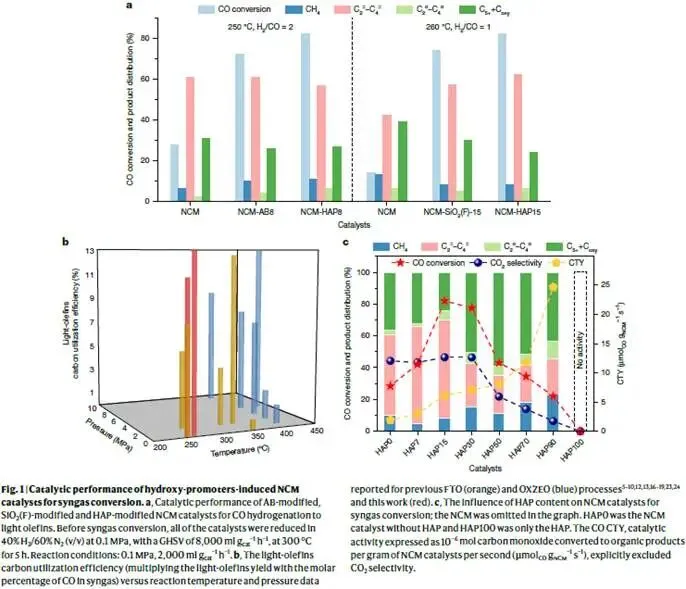
解析:图1a对比了未修饰NCM与三种羟基促进剂(AB、SiO₂(F)、HAP)物理混合后的催化性能。反应条件:250 °C、0.1 MPa、H₂/CO=2、GHSV=2000 ml·g⁻¹·h⁻¹。未修饰NCM的CO转化率仅28%,低碳烯烃(C₂⁻–C₄⁻)选择性约61%。引入AB后,CO转化率飙升至72%,烯烃选择性保持在61%;引入HAP后,转化率进一步提高至82%,烯烃选择性57%;SiO₂(F)效果介于两者之间。三种促进剂本身(如HAP100)几乎无活性,说明它们与NCM之间存在强烈的协同效应。值得注意的是,CO₂选择性在所有样品中约为45%,这是FTO路线的典型特征(水煤气变换反应副产CO₂)。然而,在H₂/CO=1的低氢碳比下,该体系仍能高效运行(见下文),意味着碳原子利用效率更高。
图1b将本研究结果(红点)与文献报道的FTO(橙色)和OXZEO(蓝色)体系进行对比,纵坐标为低碳烯烃碳利用率(定义为烯烃产率×合成气中CO摩尔百分比),横坐标为反应温度和压力。OXZEO路线虽然烯烃选择性高,但需要高温高压;传统FTO路线常在较高H₂/CO比下运行,碳利用率受限。本研究在最低温度(250–260 °C)和常压下实现了13%的碳利用率,处于领先水平,充分展示了温和条件下的优势。
图1c考察了HAP含量对催化性能的影响(H₂/CO=1,260 °C)。随着HAP从0增加到15 wt%,CO转化率从约38%升至82%,烯烃选择性从约54%升至62%,CO₂选择性保持在约45%。进一步提高HAP至30%,转化率和烯烃选择性均下降。CO CTY(单位质量NCM催化剂转化CO的速率,排除CO₂)随HAP增加而持续上升,表明羟基促进剂不仅增强了活性,还可能提高了活性位点的分散度。当H₂/CO降至0.8时(补充表5),CO转化率仍保持在64%左右,烯烃选择性约60%,展示了该策略对低H₂/CO比的适应性——这对于以煤为源头(H₂/CO≈0.5)的合成气尤为重要。
核心结论:羟基促进剂大幅提升了NCM催化剂的CO转化率和烯烃选择性,最佳添加量约为15 wt%。该体系可在宽H₂/CO范围(0.8–2)内高效运行,碳利用率创同类报道新高。
图2. 晶体结构与形貌分析
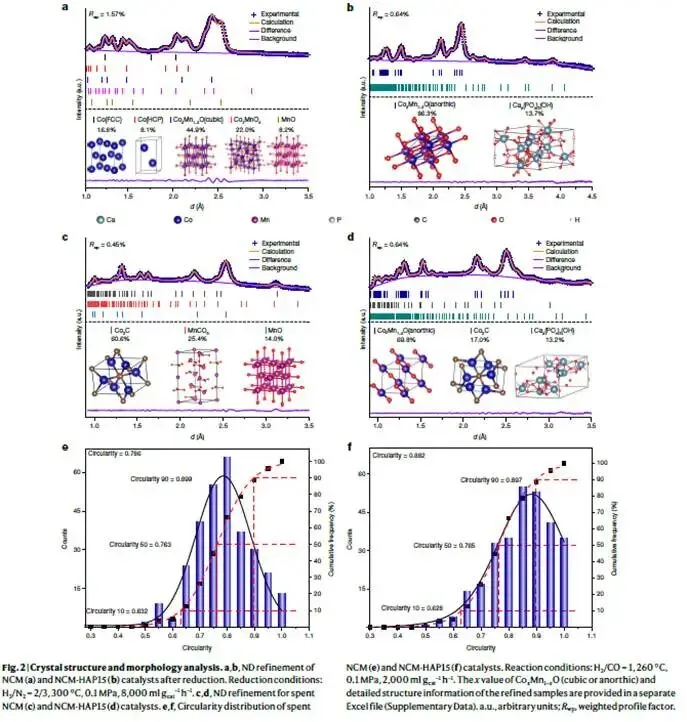
解析:图2a-b为还原后(300 °C,H₂/N₂中还原5 h)NCM和NCM-HAP15的中子衍射(ND)精修结果。中子衍射对轻元素(O、C)和相邻过渡金属(Co、Mn)的区分能力强于XRD。NCM(图2a)呈现高对称性立方相(Co-Mn氧化物),并出现明显的金属Co⁰衍射峰(箭头处)。而NCM-HAP15(图2b)则转变为低对称性的三斜晶系(anorthic)相(晶格参数见补充数据),且无Co⁰峰。这表明羟基促进剂抑制了氧化物的深度还原,并诱导了对称性降低的结构转变。H₂-TPR(补充图8a)证实,随着HAP含量增加,还原峰向高温移动,说明氧化物更难被还原。
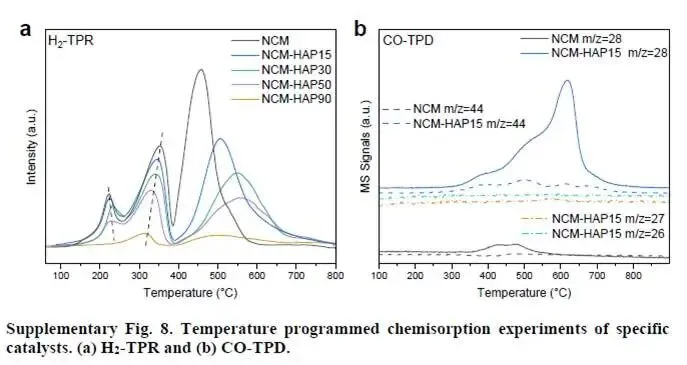
图2c-d为反应后(spent)样品的ND精修。NCM(图2c)中Co₂C占60.6%,MnCO₃占25.4%,立方MnO占14.0%。而NCM-HAP15(图2d)中Co₂C仅占17.0%,三斜相Co-Mn氧化物仍占69.8%,表明羟基促进剂抑制了碳化反应,保持了氧化物的主导地位。补充图14c显示,随着HAP含量增加,三斜相氧化物相比例升高,Co₂C比例下降,而烯烃选择性先升后降。这说明适量的Co₂C对C–C偶联至关重要,但过多则导致过度加氢和石蜡生成。因此,羟基促进剂实现了“氧化物主导活化、碳化物辅助偶联”的最佳平衡。
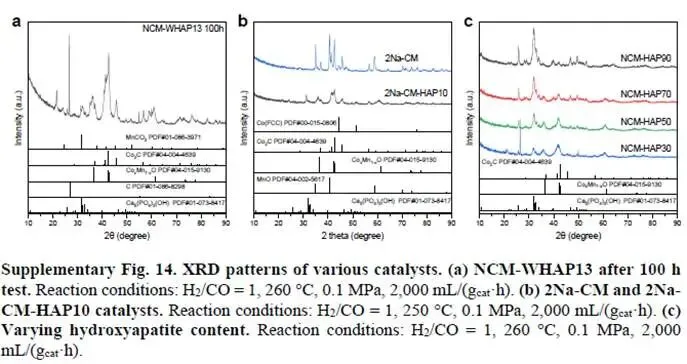
图2e-f通过圆形度(circularity)定量表征了反应后纳米颗粒的形貌。圆形度=4π×面积/周长²,完美圆为1。NCM(图2e)平均圆形度0.786,颗粒呈棱柱状;NCM-HAP15(图2f)平均圆形度0.882,颗粒更接近球形。补充图28–29的TEM图像进一步证实,NCM-HAP15的颗粒尺寸(~12.3 nm)远小于NCM(~33.7 nm),表明羟基促进剂有效抑制了烧结,并通过界面锚定稳定了小尺寸颗粒。
核心结论:羟基促进剂通过抑制还原和碳化,稳定了三斜相Co-Mn氧化物相,同时抑制颗粒长大和形貌演变,这是高性能的结构基础。
图3. 局域配位环境与表面化学态
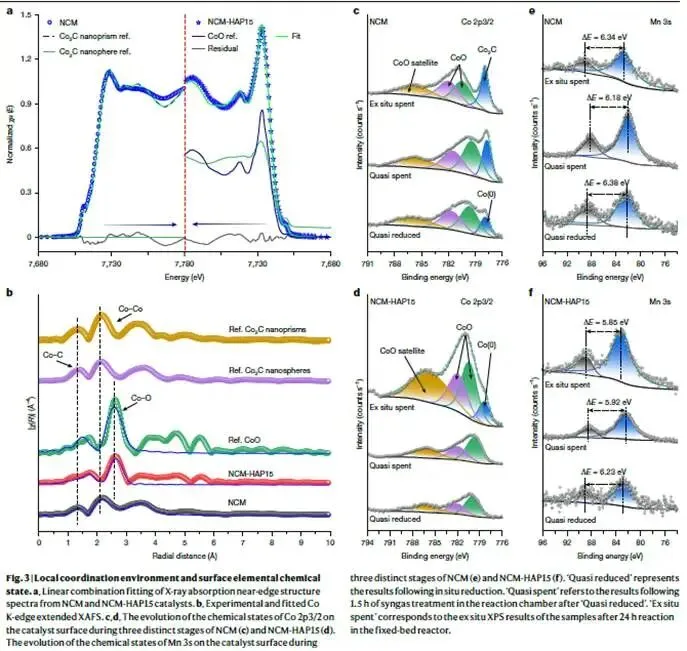
解析:图3a为反应后NCM和NCM-HAP15的Co K边XANES线性组合拟合。通过与标准样品(Co₂C纳米棱柱、Co₂C纳米球、CoO、Co箔)拟合,NCM的光谱与Co₂C纳米棱柱高度吻合;而NCM-HAP15则表现为Co₂C纳米球(51.5%)和CoO(47.4%)的混合物。这说明羟基促进剂不仅抑制了碳化程度,还改变了Co₂C的形貌(从棱柱变为球形)。棱柱状Co₂C被认为有利于烯烃生成,但本研究中NCM-HAP15的烯烃选择性并未下降,意味着烯烃生成的主要贡献可能来自氧化物–碳化物界面而非Co₂C本身的晶面效应。
图3b为Co K边EXAFS谱及拟合。NCM在1.35、2.14和2.62 Å处出现Co–C、Co–Co和Co–O散射峰,与Co₂C一致。NCM-HAP15则主要呈现Co–O特征(~2.62 Å),接近CoO。小波变换(补充图49)进一步确认了这些差异。
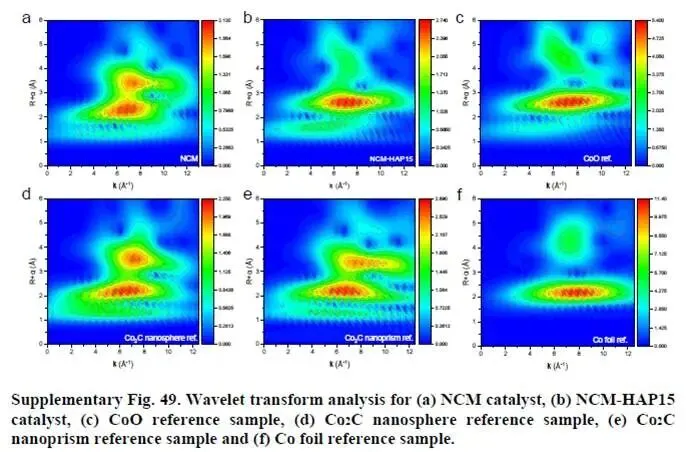
图3c-d为NCM和NCM-HAP15在不同阶段的Co 2p₃/₂ XPS谱:
- “Quasi reduced”(准还原后):NCM出现明显的Co⁰特征(~778 eV),而NCM-HAP15仍以氧化态Co为主(~780 eV)。
- “Quasi spent”(1.5 h反应后):NCM的Co⁰转化为Co₂C(~782 eV),NCM-HAP15仍保持氧化态。
- “Ex situ spent”(24 h反应后):NCM进一步碳化,NCM-HAP15仅出现微弱金属特征。
这清晰地展示了羟基促进剂全程抑制还原和碳化的动力学作用。
图3e-f为Mn 3s XPS谱。Mn 3s多重分裂间距与Mn氧化态相关。两种体系中Mn均以+2价为主,但NCM中Mn主要以MnCO₃形式存在,而NCM-HAP15中以MnO为主,与ND结果一致。
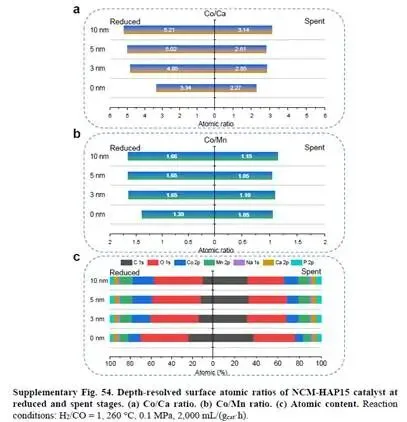
表面元素定量(补充表10)显示,NCM表面Na富集严重(Co/Na=0.6),而NCM-HAP15表面Na富集减轻(Co/Na=3.3)。深度剖析(补充图54)表明,HAP在还原阶段主要与最外层相互作用,反应期间逐渐渗透到更深层,持续调控结构。
核心结论:羟基促进剂通过界面锚定作用,抑制了Co-Mn氧化物的过度还原和碳化,稳定了三斜相氧化物相,并改变了表面Na分布和Co₂C形貌。
图4. 反应机理与理论计算
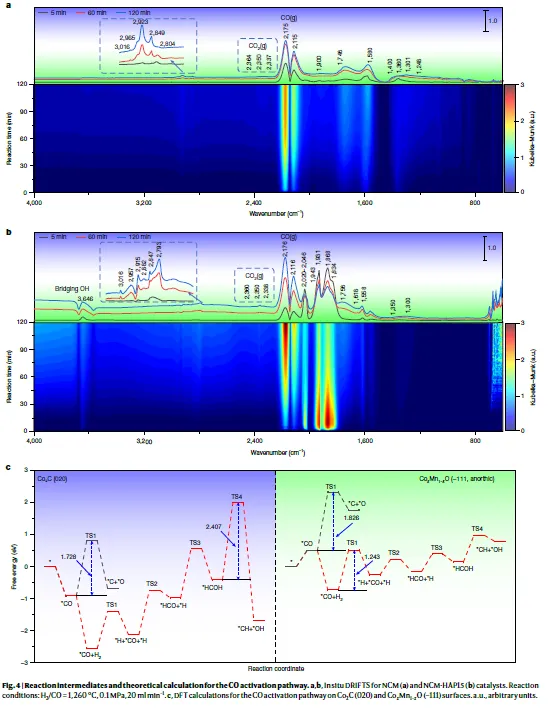
解析:图4a-b为NCM和NCM-HAP15在260 °C、H₂/CO=1条件下的原位DRIFTS谱。
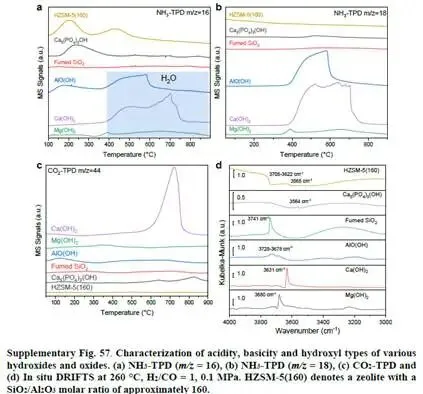
- 3646 cm⁻¹:NCM-HAP15出现明显的桥式羟基伸缩振动峰,该峰在NCM中不存在,且独立HAP或HZSM-5中也不存在(补充图57),说明该信号源于羟基促进剂与三斜相Co-Mn氧化物之间的界面。这是羟基参与催化的直接证据。
- 2700–3016 cm⁻¹(C–H伸缩振动区):NCM-HAP15在约2793 cm⁻¹处出现强醛类特征峰,指示H*辅助CO活化路径生成的醛中间体(如HCHO或CH₃CHO)。NCM中醛类信号极弱。
- 1800–2050 cm⁻¹(CO化学吸附区):NCM-HAP15表现出更强、更多样化的CO吸附峰,表明其具有更丰富的CO结合位点,这与三斜相氧化物表面存在多种配位不饱和位点一致。
图4c为DFT计算对比了Co₂C(020)面和三斜相Co₀.₅Mn₀.₅O(-111)面上直接CO解离与H*辅助CO解离的自由能垒。
- 在Co₂C(020)上:直接解离路径的能垒为1.728 eV,而H辅助路径需要2.407 eV,说明Co₂C倾向于直接解离。直接解离生成的C和O*会被快速加氢生成CH₄和H₂O,这是传统Co₂C催化剂甲烷选择性高的根本原因。
- 在Co₀.₅Mn₀.₅O(-111)上:H*辅助路径(CO吸附在Mn位点)的最高能垒为1.243 eV,远低于直接解离路径的1.826 eV。这表明三斜相Co-Mn氧化物表面**优先选择H*辅助路径**,该路径生成CHₓO中间体(如HCO、HCOOH等),这些含氧中间体迁移至Co₂C或界面后进行C–C偶联,有利于生成烯烃而非烷烃。
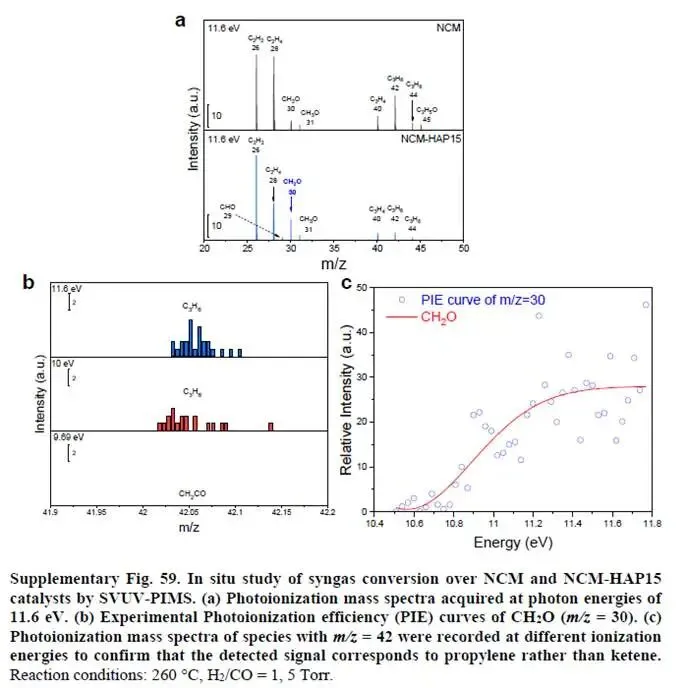
补充图59的SVUV-PIMS(同步辐射真空紫外光电离质谱)检测到NCM-HAP15反应气相中m/z=30(CH₂O异构体)信号明显高于NCM,直接证实了H*辅助路径中间体的存在。
核心结论:羟基诱导的三斜相Co-Mn氧化物通过H*辅助CO活化路径,在较低能垒下生成含氧中间体,这些中间体在Co₂C或氧化物–碳化物界面发生C–C偶联,高效生成低碳烯烃。这一机制从根本上解决了传统FTO中直接解离路径导致的过度加氢问题。
总结与展望
本研究通过引入羟基促进剂与Co₂MnO₄前驱体物理混合,开发了一种全新的合成气制低碳烯烃催化体系,主要结论如下:
- 催化剂结构:羟基促进剂诱导Co-Mn氧化物从高对称立方相转变为低对称三斜相,抑制了氧化物的过度还原和碳化,稳定了三斜相Co-Mn氧化物作为主要活性相,同时生成适量Co₂C负责C–C偶联。
- 反应机理:三斜相Co-Mn氧化物通过H*辅助CO解离路径(能垒1.243 eV)活化CO,生成CHₓO等含氧中间体,然后迁移至Co₂C或界面进行C–C偶联生成烯烃。这避免了直接解离路径(能垒>1.7 eV)伴随的深度加氢和甲烷生成。
- 催化性能:在250–260 °C、0.1 MPa、H₂/CO=1–2的温和条件下,实现了70–82% CO转化率、>60%低碳烯烃选择性,碳利用率高达13%,处于同类报道领先水平。催化剂在100小时运行中保持稳定。
- 普适性:该策略适用于多种羟基供体(Ca、Si、Al基)、不同碱金属(Na、K)和不同Co/Mn比例,展示出良好的通用性。
科学意义:本研究首次提出了“羟基诱导三斜相钴锰氧化物”作为CO活化新活性相的概念,揭示了H*辅助CO解离路径在温和条件下合成气制烯烃中的优越性。这一发现不仅为FTO催化剂设计提供了全新思路,也为其他涉及CO活化的反应(如CO₂加氢、甲烷重整)提供了借鉴。与前期疏水策略(促进H₂O脱附)相比,本研究的亲水策略(利用羟基稳定氧化物相)形成了原理性互补,展示了界面化学调控的丰富可能性。
未来展望:该策略有望进一步拓展至其他过渡金属氧化物体系(如Fe、Ni基),并探索在更高压力下的性能优化。深入理解羟基促进剂与三斜相氧化物之间的界面相互作用机制(如原位光谱和理论计算结合),将有助于理性设计更高效、更稳定的工业催化剂。此外,将该策略与先进的反应器工程(如膜反应器、微反应器)结合,有望进一步提升碳效率和烯烃收率。
通讯作者简介
葛庆杰(Qingjie Ge)博士,研究员。1993年毕业于郑州大学化学系, 1998年获中科院兰州化物所博士学位, 1998-2000年大连化物所博士后, 出站后留所工作, 期间于2000-2001年在法国斯特拉斯堡路易-巴斯德大学做访问学者、博士后研究。2005年-2008年, 日本北九州市立大学环境工学部特任研究员、特任教授,同时受聘于日本气体合成公司主任研究员。2008年回所工作, 担任烃类选择氧化课题组副组长、创新特区组组长,主要进行与合成气中枢相关的催化新过程和新材料的研究与开发,在Nature 等期刊发表论文多篇。
孙剑(Jian Sun)男,博士,研究员,博士生导师,研究组组长,国家青年科学基金A类项目获得者。在山东大学获工学学士和硕士学位;2015年毕业于日本国立富山大学应用化学系,师从椿范立院士,获得博士学位,同年以海外引进人才加入中国科学院大连化学物理研究所,先后任副研究员、项目研究员,2022年晋升为研究员。围绕二氧化碳催化转化方向开展研究。带领团队首创了二氧化碳加氢制绿色油品的新过程,被《Nature》杂志选为研究亮点。完成全球首套年产1000吨二氧化碳加氢制汽油工业示范,科技成果被石化联合会评价为国际领先水平。以通讯作者在Nature、Nat. Chem., Nat. Synth., Nat. Commun., Sci. Adv., J. Am. Chem. Soc., Angew. Chem., Chem. Soc. Rev.等权威刊物发表论文百余篇,单篇最高他引逾1000次。主持国家自然科学基金重大研究计划、国家重点研发计划等十余个项目或课题,实现技术许可或转让5000余万元。获大连最美科技工作者称号,入选全球前2%顶尖科学家。以第一完成人获中国石油和化学工业联合会科技进步一等奖(2023)、中国石油和化学工业联合会青年科技突出贡献奖(2023)、侯德榜化工青年科技奖(2022)、辽宁省自然科学二等奖(2022)。研究方向:(1)二氧化碳加氢合成燃料与高值化学品;(2)合成气转化新途径;(3)纳米催化剂的设计与绿色合成。
声明:文章内容仅代表本人观点,本文数据来源于Nature论文(Nature 2026, DOI: 10.1038/s41586-026-10204-4),图片版权归原作者和出版商所有。如有侵权,请联系后台,感谢支持,欢迎批评指正!
欢迎各位老师同学们在公众号上投稿分享课题组研究成果,不限期刊和发表时间!后台联系即可。