 夜雨聆风
夜雨聆风





分子对接分数高,就说明结合好吗?
这件事,恰恰是很多文章和项目里最容易被误解的地方。
一、为什么大家会误以为“分数高 = 结合好”?
因为分子对接本来就是用来做“构象预测”和“初步排序”的。它会把小分子放进蛋白口袋里,生成多个可能的结合姿势,再用评分函数去打分,最后给出一个“看起来最优”的结果。
所以从使用习惯上看,大家很容易觉得:
-
分数更优,说明结合更稳定
-
分数差得多,说明两个分子差异很明显
-
最低能构象,就是“真实构象”
但问题在于,评分函数本身是简化模型。它并没有真正把结合过程中的所有物理化学因素都完整算进去。尤其是蛋白柔性、溶剂效应、熵效应、诱导契合这些因素,往往处理得并不充分。
所以,对接分数有参考意义,但不能被当成“实验真值”。
二、对接分数到底代表什么?
对接软件给出的分数,本质上是一个经验化、近似化的评价值。它的作用更接近于:“在这一批候选构象里,程序觉得哪几个更像是合理结合模式。”注意,是“更像合理”,不是“已经证明结合一定更强”。
以 AutoDock Vina 为例,它的评分函数是基于已有复合物数据集和经验项建立的,主要目的是在速度和准确性之间做平衡,用于姿势筛选和相对排序,而不是严格替代实验测得的结合自由能。
所以在实际工作里,对接分数更适合这样理解:
- 适合做初筛
- 适合在同一体系内做相对比较
- 适合帮助筛选可能的结合模式
- 不适合单独拿来证明“这个分子一定结合更强”
三、为什么“分数高”不一定真的是“结合好”?
1)评分函数是简化的,不是完整自由能
真实结合过程里,除了静态相互作用,还涉及:
-
蛋白构象变化 -
配体构象代价 -
水分子重排 -
离子环境影响 -
熵损失 -
诱导契合效应
而很多常规 docking 打分函数,对这些因素只是粗略处理,甚至基本没有完整体现。
所以,分数只是“近似评价”,不是严格意义上的实验结合自由能。
2)蛋白往往不是刚性的
很多对接默认受体基本刚性,最多允许部分侧链微调。但现实里,蛋白口袋常常会“动”,有些结合甚至依赖明显的 loop 变化、侧链翻转或诱导契合。如果蛋白真实构象和你拿去 docking 的那一个差别较大,分数再好,也可能只是“在这个静态结构上看起来不错”。
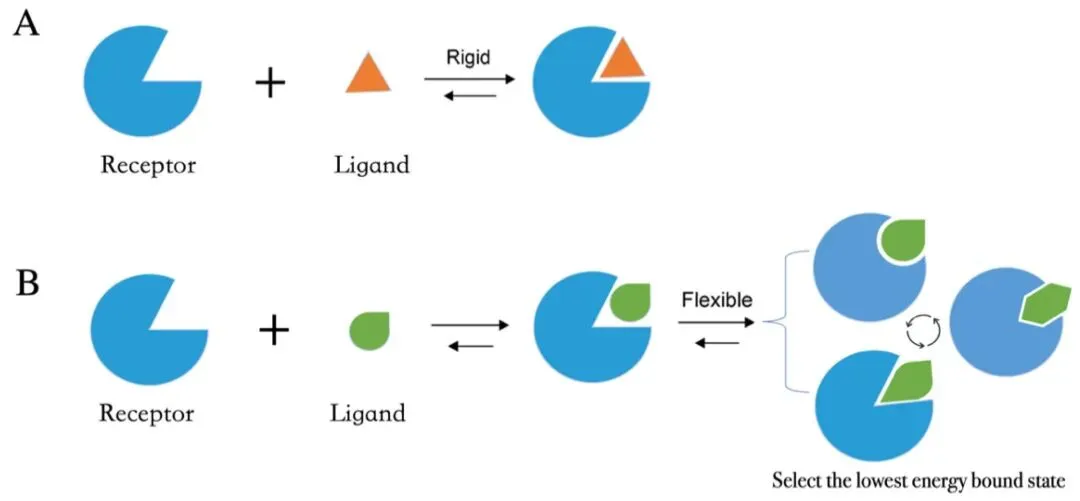
图1:刚性对接 vs 柔性对接示意图
3)大分子、疏水分子有时容易“占便宜”
某些 docking score 与分子大小、非特异性接触有一定相关性。换句话说,分子更大、接触面积更大,有时会更容易拿到“看起来不错”的分数,但这并不一定意味着真正更特异、更有效。
4)分数接近时,通常不能过度解读
比如两个构象一个是 -7.8 kcal/mol,一个是 -7.5 kcal/mol。这种差异在很多情况下其实并不算大,未必足以支持“前者明显优于后者”的结论。因为 docking 本身存在采样误差、评分误差和建模误差。
四、那对接结果到底该怎么看,才算比较靠谱?
第一,看口袋是否合理:分数再好,如果对接位置不对,意义也不大。要先确认:
-
是不是落在已知活性口袋/结合位点 -
是否和已知关键残基有对应关系 -
是否符合文献已报道的作用区域 -
是否和共晶配体位置一致或接近
也就是说,先看“对没对地方”,再看“分高不高”。
第二,看结合构象是否讲得通:一个可信的 docking pose,通常应该在结构上是“能解释得通”的,比如:
-
有明确的氢键锚定 -
疏水骨架嵌入口袋 -
带电基团位置合理 -
没有明显穿模、严重碰撞或不自然伸展
对接不是只看一个数字,而是要看这个构象是否“像真的”。
第三,看关键相互作用能不能自洽:通常会重点看:
-
氢键 -
疏水作用 -
π-π / π-cation 相互作用 -
盐桥 / 静电作用 -
金属配位(如果体系里有金属)
如果分数很好,但关键相互作用解释不出来,这种结果往往不够扎实。
第四,看是否做过对接验证:一个相对规范的 docking 流程,通常会做 redocking或方法验证。也就是把共晶配体拿出来重新对接,看程序能不能把它放回接近实验的位置。很多研究里会把 RMSD < 2 Å作为一个常见的成功参考标准。这一步很重要,因为它能说明:你的参数设置、网格范围和对接流程,至少对这个体系不是“完全飘的”。
第五,看后续动力学是否稳定:这也是为什么很多研究做完 docking 以后,还要接着做 MD。
因为 docking 给你的只是一个静态瞬间;而分子动力学更关注的是:
-
这个构象在时间尺度上稳不稳 -
关键氢键保不保得住 -
配体会不会很快跑掉 -
蛋白口袋会不会发生重排 -
结合界面是不是能持续维持
所以,docking 更像“先找到可能的姿势”,MD 才是在看“这个姿势站不站得住”。
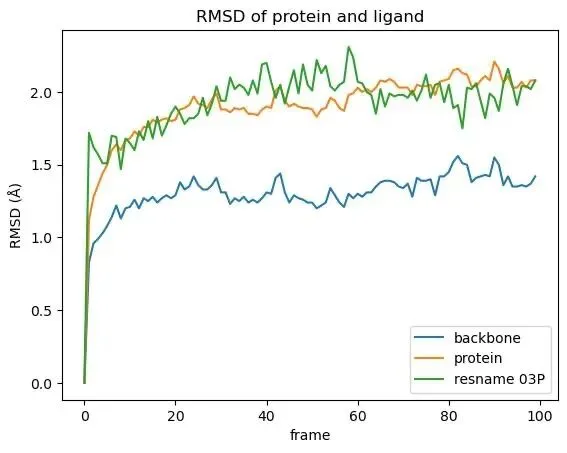
图2:MD 里的 RMSD 图
五、如果是蛋白-蛋白对接,思路有什么不一样?
前面讲的内容,主要更适用于蛋白-小分子对接;但如果是蛋白-蛋白对接,也同样不能简单理解成“分数高就一定结合更好”。因为蛋白-蛋白体系通常更复杂,界面更大,也更容易受到构象变化影响。在这种情况下,除了分数,更应该重点看:
-
对接界面是否落在已知或合理的互作区域 -
界面残基是否能解释关键氢键、盐桥和疏水接触 -
两个蛋白的空间排布是否合理,有没有明显冲突 -
界面面积和接触网络是否足够连续 -
后续 MD 中界面是否还能维持稳定
也就是说,蛋白-蛋白对接里,分数通常更不能单独拿来下结论。真正有说服力的,往往是“界面位置 + 关键残基 + 接触网络 + 动态稳定性”一起看。
图3:蛋白蛋白互作展示图,蛋白-蛋白不能只看分数,更要看界面位置、关键残基。
六、哪些情况下,对接分数特别不能单独信?
下面这几种情况,尤其要小心:
1)蛋白口袋很柔性:比如 loop 很多、诱导契合明显的体系。这种情况下,刚性 docking 往往很容易失真。
2)有金属离子、辅因子、结构水参与结合:这类体系对参数和相互作用模型要求更高。如果处理不好,分数很容易看起来漂亮,但机制并不可靠。
3)比较的是差异很小的一组分子:这种情况下,score 的细微差异往往不足以支撑强结论。更适合结合相互作用分析、MD、MM/PBSA 或实验一起看。
4)想直接拿 docking score 替代实验亲和力:这个是最常见误区。常规 docking 方法通常并不可靠于直接预测真实 binding affinity 的绝对值。
七、一个更实用的判断思路
如果你真的要判断“这个分子是不是更可能结合得好”,建议不要只盯着分数,而是按下面这个顺序看:
-
位置对不对,是不是在合理口袋里,是否符合已知位点信息。
-
构象顺不顺,有没有明显穿插、冲突,不合理暴露,或关键基团朝向错误。
-
作用合不合理,能不能解释出关键氢键、疏水锚定、静电作用、金属配位等。
-
验证做没做,有没有 redocking,参数是否经过基本验证。
-
动态稳不稳,有没有 MD 支撑,关键作用是否可持续存在。
-
如果是蛋白-蛋白,对界面是否讲得通,界面是不是合理,关键残基和接触网络是否能够自洽,后续界面是否稳定。
-
能否和实验或文献对上,有没有突变实验、活性数据、SPR/ITC、已知 SAR 或共晶结构来支撑。
只有把这些放在一起看,对接结果才更有说服力。
八、写论文或写报告时,更稳妥的表达方式是什么?
不要直接写:“该配体与蛋白结合能力最强。”更稳妥的写法是:
- “对接结果提示该配体在该位点具有较优结合倾向。”
- “该构象显示出较有利的相互作用模式。”
- “在当前对接模型下,该分子表现出相对更优的评分和更合理的界面作用。”
- “结果提示其可能具有较好的结合潜力,但仍需结合分子动力学模拟或实验进一步验证。”
如果是蛋白-蛋白体系,也可以写:
- “蛋白-蛋白对接结果提示两者可能通过该界面发生相互作用,但其稳定性仍需结合界面分析和后续动力学模拟进一步评估。”
这样的表述会专业很多,也更不容易被审稿人或同行挑问题。
九、总结:对接分数能看,但不能只看
回到最开始那个问题:对接分数高,就说明结合好吗?答案是:不能直接这么下结论。更准确地说,高分只能说明它在当前 docking 模型里“更被看好”,但是否真的结合更强,还要结合口袋合理性、构象质量、关键相互作用、界面分析、方法验证、分子动力学以及实验证据综合判断。
这也是为什么,真正靠谱的计算分析,往往不是只报一个分数,而是要把“分数 + 构象 + 作用模式 + 动态稳定性”一起讲清楚。